随着雾霾话题的讨论越来越深入,关于医疗实践的探讨不绝于耳。
海普洛斯联合创始人兼CTO陈实富在云栖TechDay的分享非常有意义,聚焦如何利用互联网+基因技术进行液体活检和精准分析,进而改变肿瘤的诊断与治疗。陈实富是NVIDIA高级工程师,GPU计算专家,目前主攻肿瘤基因测序和数据分析,申请有十余项专利,并负责多项深圳市技术攻关项目,也是开源基因数据分析项目OpenGene的发起人。

我们可以说互联网是我们生活的一个基础设施,而基因则会是我们健康的一个基础设施。刚刚泽辉也说了,生命是可以数字化的,其实数字也可以生命化,数字会有生命,这就是我们常说的人工智能。今天我们讲讲,生命数字化与数字生命化两个里面一个交汇处,就是我们的医学,医学里面很重要的一部分就是肿瘤学,怎么样通过基因方面的技术,去改变肿瘤的诊断和治疗?今天我们聊聊一个方向,即肿瘤的液体活检,以及说说怎么通过我们分析的方法精准地去分析肿瘤的信息。

很久以来,我们一直都在寻找,有没有更好的方法可以去诊断癌症,因为癌症在很早期被发现的时候它只是一个常规疾病,可以被切除被治愈的。但是到了晚期的时候就是一个绝症,那我们如果能够准确快速简单地去测量癌症,我们就有办法可以把它治愈。我们知道,肿瘤其实是一种基因疾病,是基因突变的产生、积累导致细胞不能被修复,如果细胞不能被修复而且不能被清除,就会不断的积累,不断的恶性增殖,直到形成一个肿块,就是一个肿瘤。所以往往是先有基因突变产生,然后才会有肿瘤。那如果能够获取这个细胞里的基因信息,我们就可以对肿瘤可以有一个全面的了解。

但是很长时间以来,我们都靠显微镜去看,因为为什么呢?因为我们没有更好的方法去检测细胞里的DNA的信息。
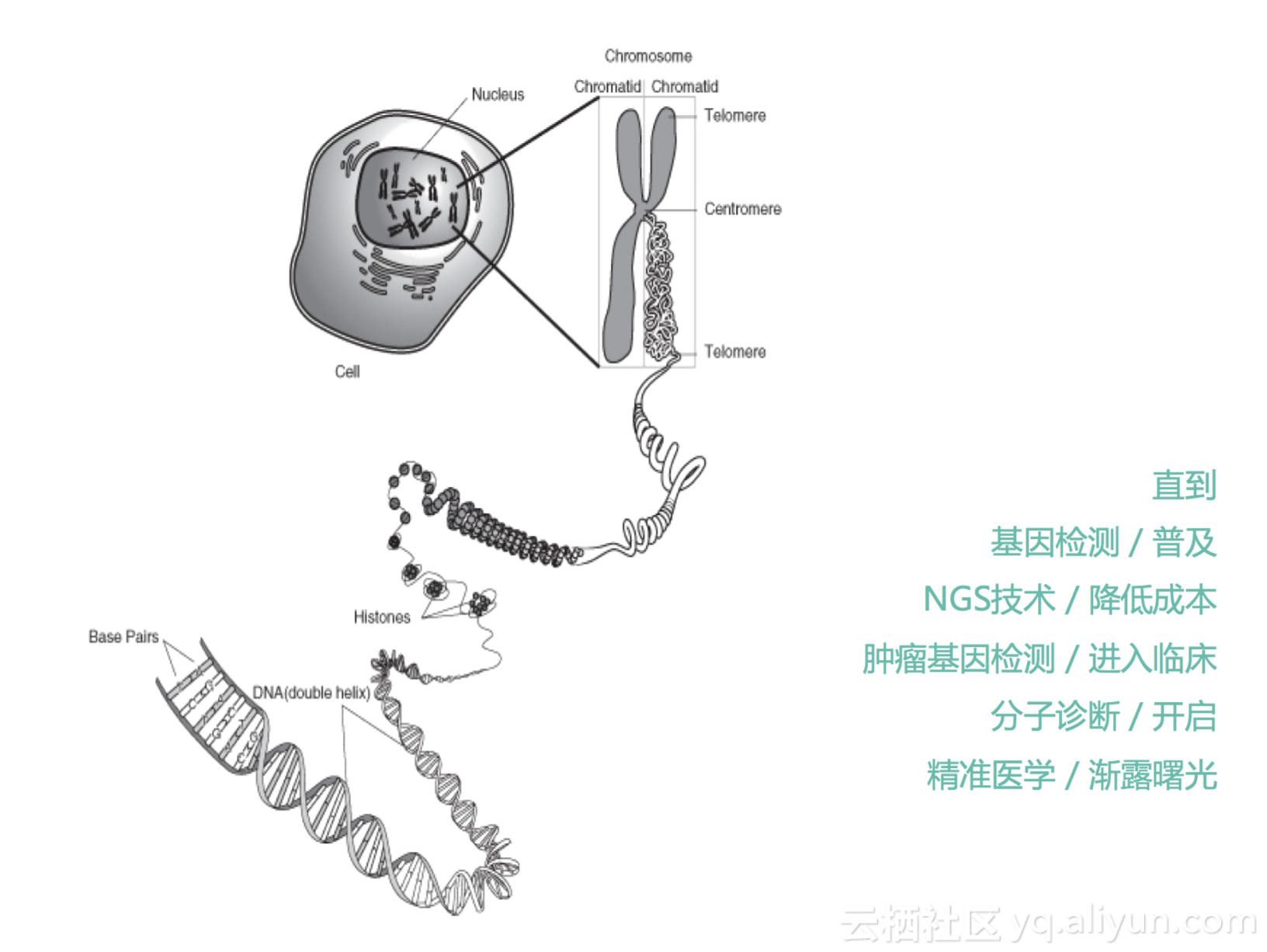
直到我们后面有了基因测序,我们可以通过基因测序的方法可以去检测我们细胞里面的这些信息,我们细胞里面有23对染色体,可以通过我们的测序技术,去获取里面DNA序列的信息。特点地,对于肿瘤细胞来说,它就会带有我们肿瘤细胞一些特异的变异,了解这些肿瘤的基因变异是十分关键的。
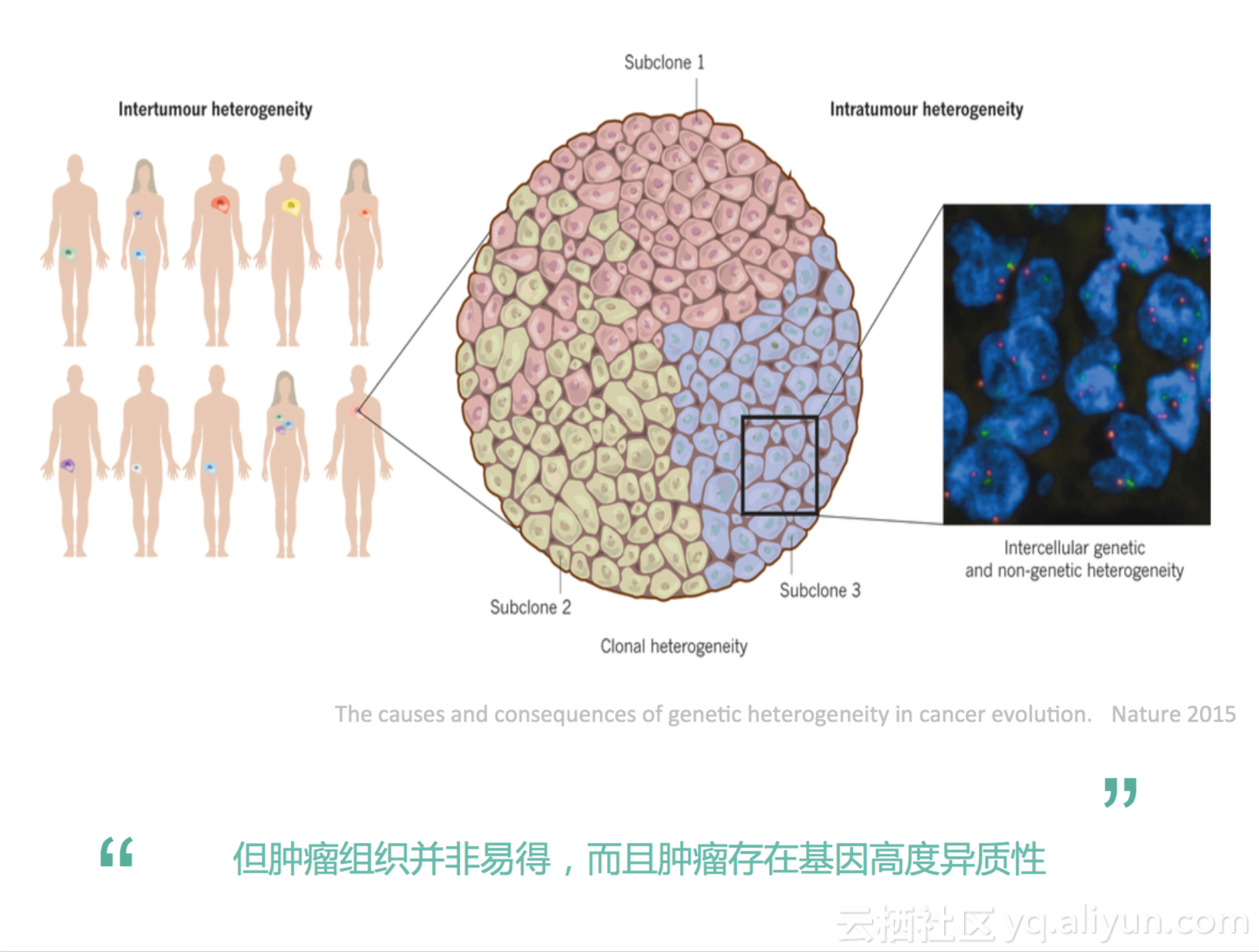
但是我们传统的方法是有一些问题,为什么呢?我们一般是通过一个手术活检可以获取一些组织,这块组织我们可以进行基因检测,然后进行分析它里面有什么基因变异。但是,并不是每一个患者都有条件进行活检的,因为我们去做活检的时候是有创伤的,很多患者他在晚期的时候是没有办法取这个活检的。有人说我们去做穿刺,穿刺样本的话,也是有损伤,比如说有的肺癌患者,他晚期的时候不敢穿刺,穿刺有风险。另外一个问题是什么呢?肿瘤它有异质性,不同人的肿瘤会有异质性,同一个人的肿瘤在不同位置有异质性,即便是同一个人,同一个位置肿瘤也会有很多亚克隆,这是个肿瘤组织。这一块跟那一块还是不一样的。即便是同一个克隆体,它里面每一个细胞也是不一样的。你看这个图,细胞里面有绿色有红色的,它们发的是不一样的光,表示它在DNA或者蛋白层面上是很不一样的,所以即便是同一个肿瘤组织里面,即便是同一个亚克隆里面每一个细胞都可以是不一样的。如果我们只是取里面的一小块去测序的话,它是没有办法去获取肿瘤全面信息的。特别是如果我们做穿刺,穿刺的时候我们只穿到里面一小块组织,可能运气好穿到了肿瘤组织,也可能运气不好穿到了癌旁组织,如果是癌旁组织,我们就不能准确地获得癌症的基因信息。

所以我们就要有新方法,什么新方法呢,那就是液体活检。就是通过一管血,就可以做到无创全面实时的癌症基因检测。什么意思呢?因为我们的癌症组织它里面会有一些细胞会死亡或者凋亡,然后里面DNA会破碎并渗透到我们血管里面去。
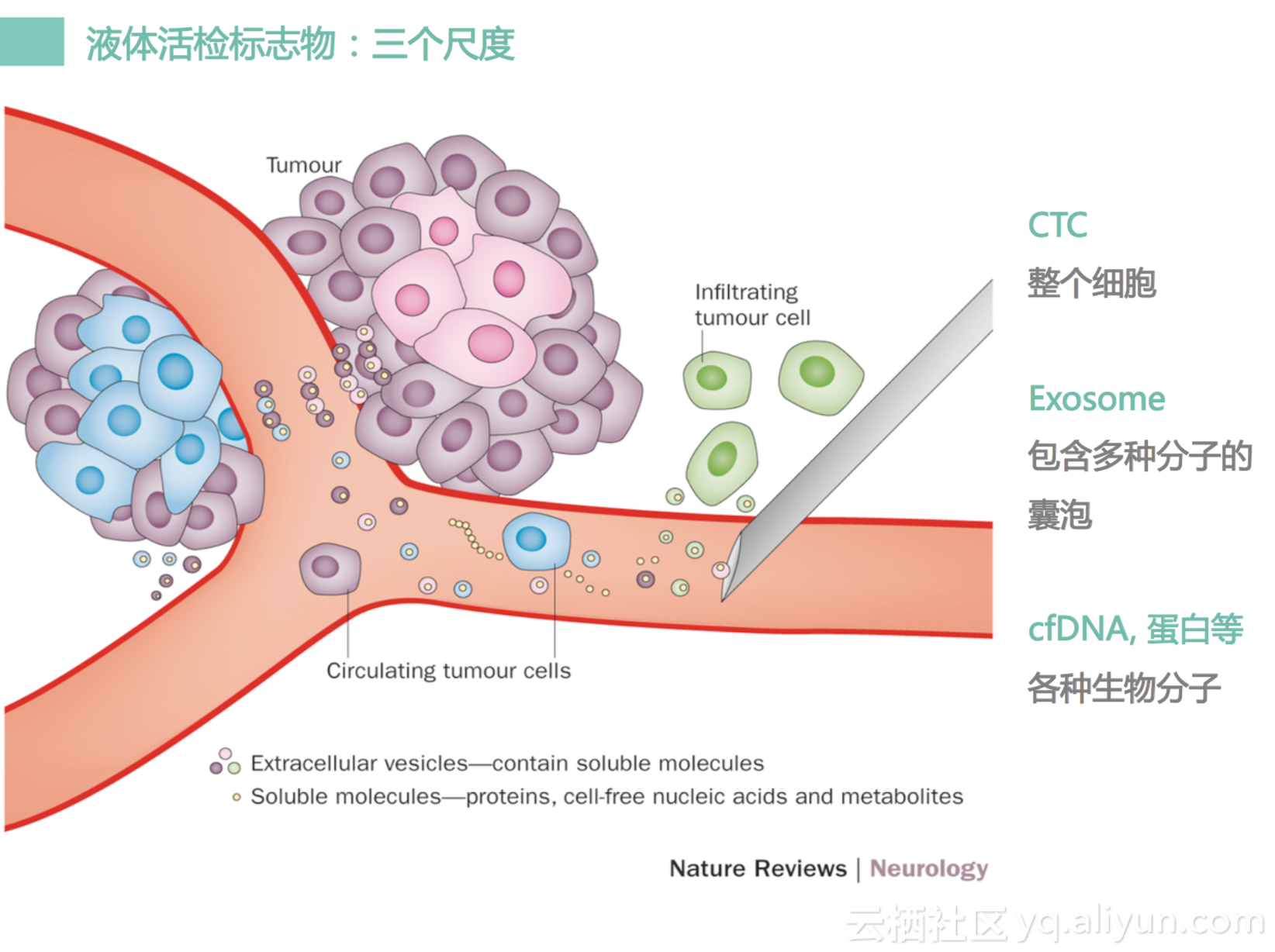
我们一般说有三个层次的液体活检:第一个层次,我们称之为CTC,就是循环肿瘤细胞,这个是整个细胞在血管里面循环,有可能是一团一团的细胞。这个细胞是有活性的,是可以做培养的。第二个层次,我们称之为Exosome,外泌体,就是我们有一些细胞内部会分泌一些囊泡,这个囊泡就带有一些核酸,包括我们DNA,还有很多RNA,还有蛋白质,这个小囊泡可以穿过细胞膜,跑到细胞外面来,成为一个游离状态。所以我们称他是一个囊泡状,它是一个运输很多大分子囊泡。然后第三个层次,是一个游离大分子,主要是有一些核酸,包括我们cell free DNA,同时还会有一些蛋白质,还有一些RNA,所以三个层次我们都可以简称,我们称为液态活检。液态并不指的是血液,也可以是汗液,也可以尿液,也可以是脑脊液,或者是腹水,胸水,甚至还可以是口水,所以就是说其实它是一个很广的概念,但是最准确最稳定的方法还是用血液。因为血液是比较容易获取,同时里面的DNA量比较多,也比较容易富集,方法也比较简单一点。
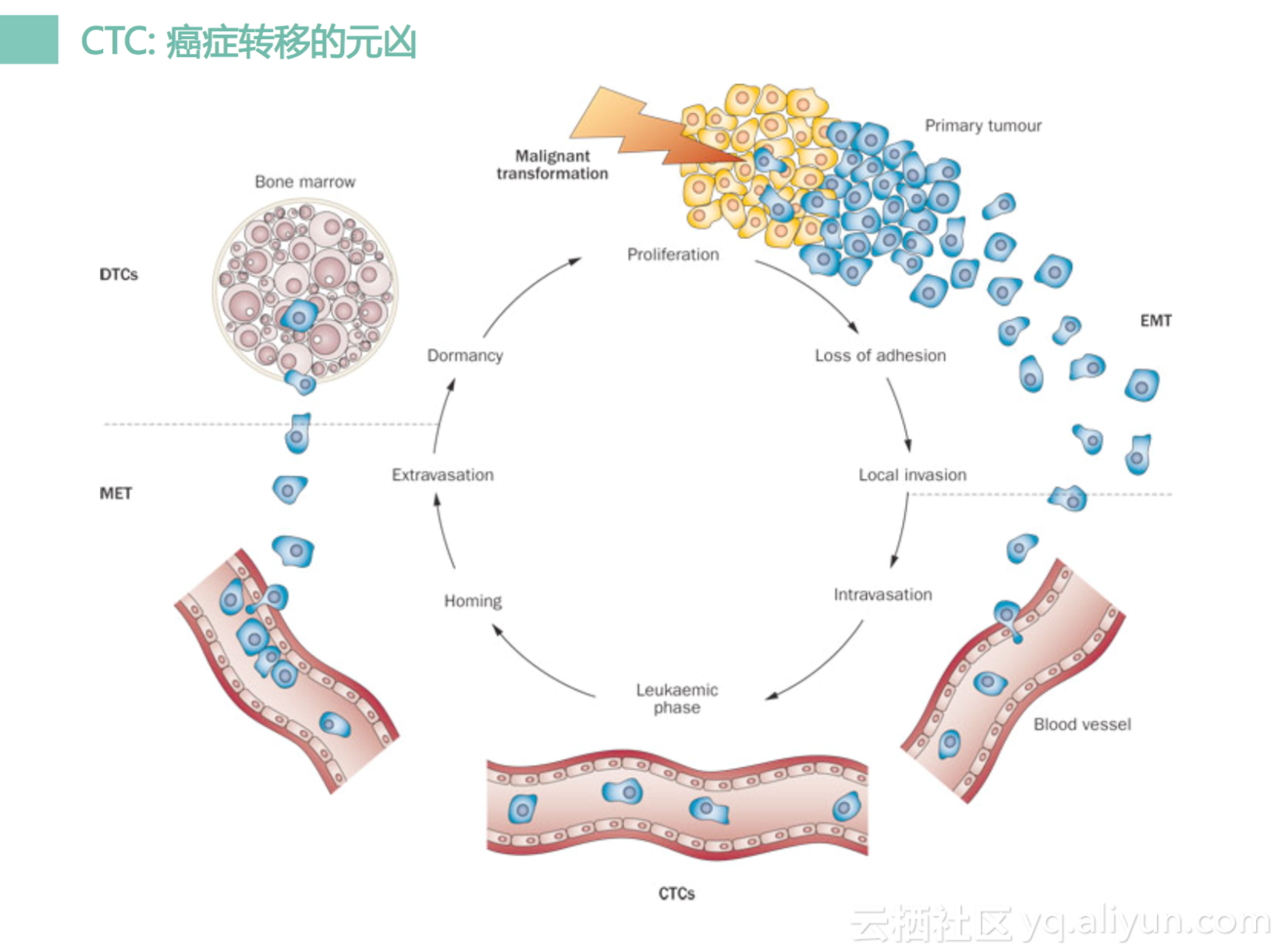
CTC可以被认为是癌症转移的元凶,为什么呢?因为肿瘤在完成近段的进入之后,它会有一些细胞脱落,脱落之后会跑到远端去,不管是经过血管或者是经过淋巴循环,跑到远端去,它就是一个种子,遇到一块合适的土壤之后,它会生根发芽,形成一个转移杜。特别是肿瘤干细胞的脱落循环更容易形成转移杜。如果能够去检测它,分析它的话,它是比较有价值的。

CTC的话,我们有几个应用,第一个的话是可以计数,我们可以算这个数目。比如说我们去抽十毫升的血,那那里面的血我们去算CTC的数目,如果我们识别到超过了10个CTC,一般这个人就可能是一个肿瘤患者,如果是低于2个一般都是良性的。然后我们可以去做测序,测序的话我们一般是把这个CTC捣碎了,为什么?因为我们很难获得很纯的CTC,一般都是CTC混合一些白细胞,因为白细胞很难被清除干净。然后我们还可以对CTC做单细胞测序,我们可以单个的CTC分离开来,分离完之后就做测序。这时候我们获得是一个纯粹的单个细胞的水平DNA或者是RNA信息。但是这个方法会特别特别困难,特别特别复杂,而且很不经济,很贵。同时我们还可以做体外培养,比如说我们可以把这个患者的CTC拿过来之后,放体外培养,然后培养完之后我们可以做药敏实验,可以把一些药物去测试它,这个细胞对这个药物会不会敏感。如果会敏感可能会预示着这个患者也可以吃这个药进行治疗。其实这个也是挺好的方向。但是当前来说的话,因为后面三个都不太成熟,不太好用,还是以CTC计为主。
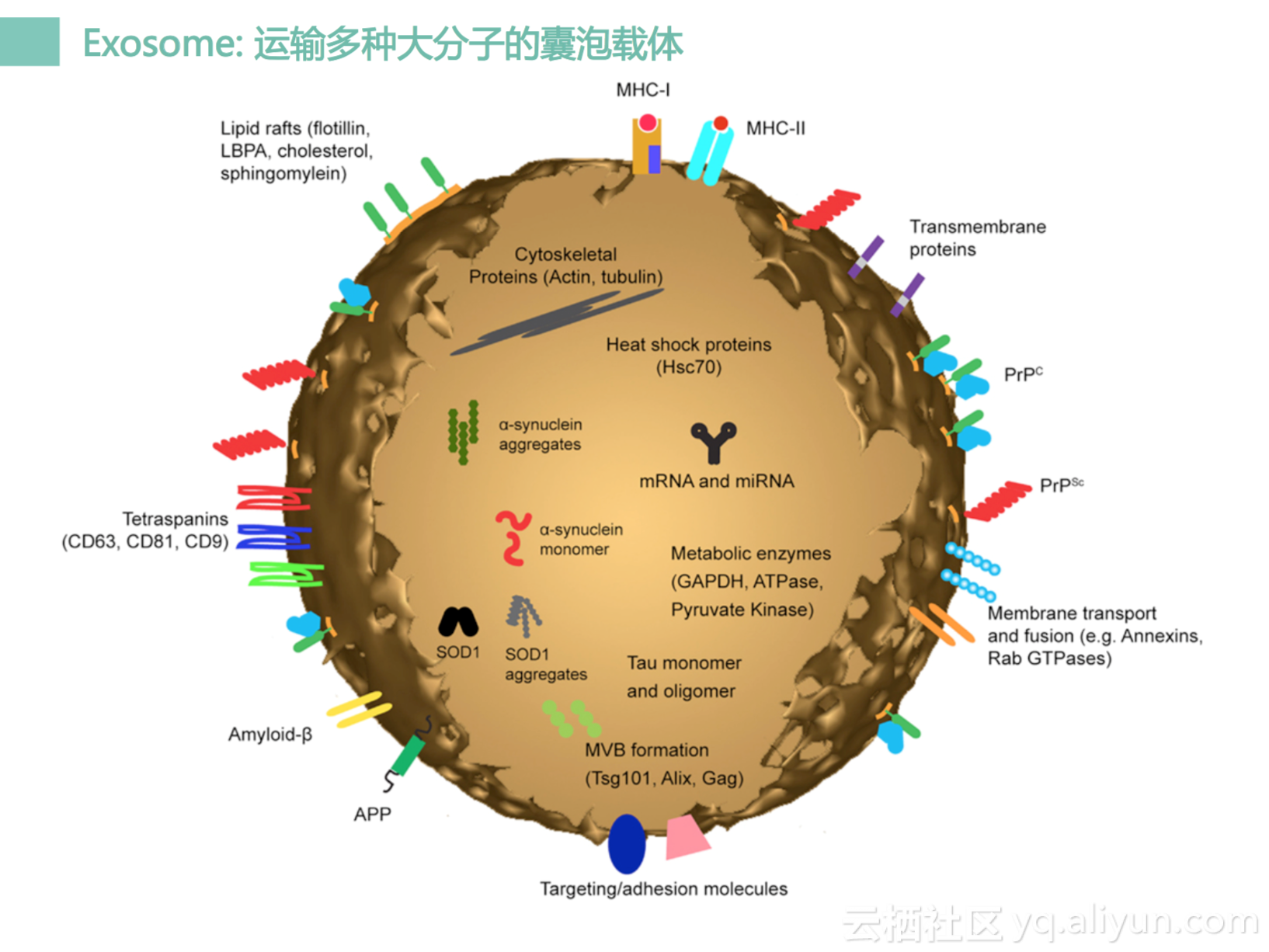
然后是外泌体,外泌体其实它是一个大囊泡,它里面有一些核酸,核酸大部分是RNA。同时它表面会有很多抗原,所以它其实是一个的杂合体,它比细胞小很多,但是一比单个DNA分子,RNA片断也会大很多。这个里面会有很多很多的信息,但是会很难分离。
它也可以测序,可以测DNA, 也可以测RNA, 一般是RNA比较多。因为有很多的蛋白质,所以也可以进行蛋白水平的分析。所以四个方面都可以做分析,但是这四个分析都很难做,因为它很难分离,所以当前只是存在于学术界,还很少有工业界来做这个事情。
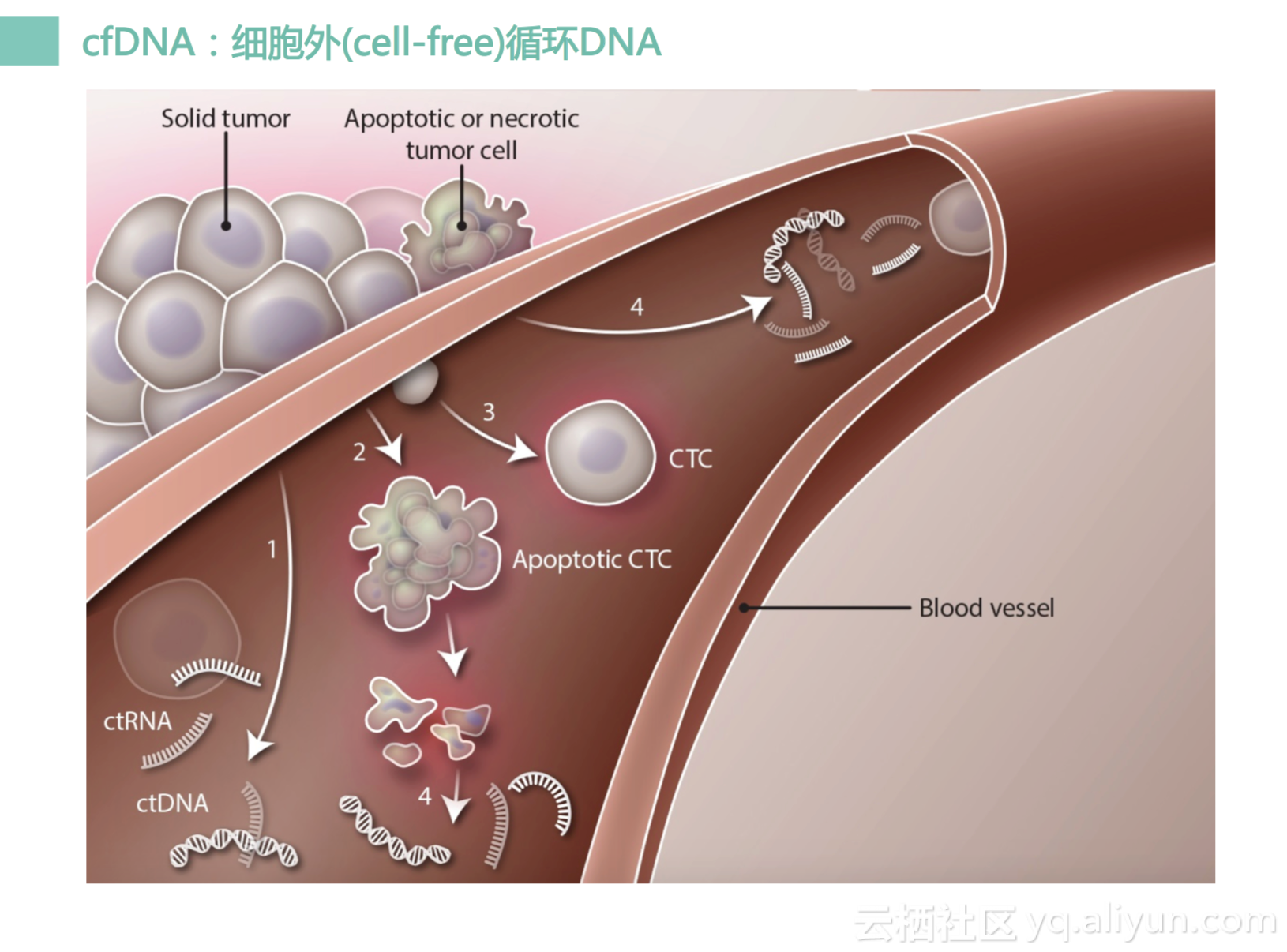
然后就是我们专门做的细胞循环DNA,就是cell-free DNA,我刚说过了肿瘤会有细胞会凋亡,会有死亡,会有DNA裂解,之后会跑到血管里面,它会形成单链DNA或者是双链游离DNA,也会有一些单链的RNA。捕获住这些DNA,我们就可以做测序。测完序之后我们就可以知道原来这个肿瘤有这样一个变异,这个变异就可以被我们识别出来,通过对他的分析,我们就可以知道这个患者是不是可以吃这个靶向药物,有没有效果,有没有什么风险。

所以我们只要能够对这个cell-free DNA进行测序的话,我们可以知道患者能吃什么药,这是个体化用药的分析,同时我们可以检测肿瘤它是什么样状态。比如说我们发现肿瘤变异,变高了,变多了,这可能是一个恶化的情况,也可能是一个转移的情况。如果它变低了,变少了,也可能是一个变好的情况。它可以做肿瘤辅助诊断,什么意思呢?有很多时候我们通过一般的方法是很难区分一个肿瘤良恶性的。比如说有一些,特别是在北方比较多,有很多人会患有肺部的结节。那这个结节的话有时候不大不小,可能一厘米左右,那通过CT看到它的话是一个病变,但是你不能分他们是良性还是恶性,然后我们一般会去拍一个PET-CT,看看这个部位的糖代谢信息,看看SUV值怎么样。SUV值如果大于5的话,一般都是一个恶性,小于5一般都是良性,但是有的时候都很模糊,可能在于4和8之间,很难区分良恶,这个时候还可以做一个纤维支气管镜。纤维支气管镜也不一定能够确诊,为什么呢?我有一个朋友也是这样子,也是一个肺癌,但是他做的时候他发现纤维支气管镜里面看起来是一个良性病变,但是他通过细胞刮片出来看又像是一个恶性,所以通过这个也很难区分到底是良性还是恶性,最后测了一个序。测完序之后,发现有一个KRAS G12V的突变,这个突变一般来说跟肿瘤关系是很大的,所以我们可以推断是一个恶性肿瘤。 然后去化疗表示有效果,可以证明确实是一个恶性肿瘤。
ctDNA还可以做筛查,就是说我们可以在肿瘤很早很小的时候,可以通过ctDNA把它测出来。因为我们通过影象学,就是我们做CT,做核磁共振等都只能够发现大概是7个毫米以上的肿块。对于一个比较小的,比如说三毫米,四毫米的话,我们没有办法发现,但是这个时候已经在血液里面有肿瘤的游离DNA存在,我们只要把DNA测出来去分析它的话就可以把他更早期定位出来,所以基于这个,我们可以做筛查。
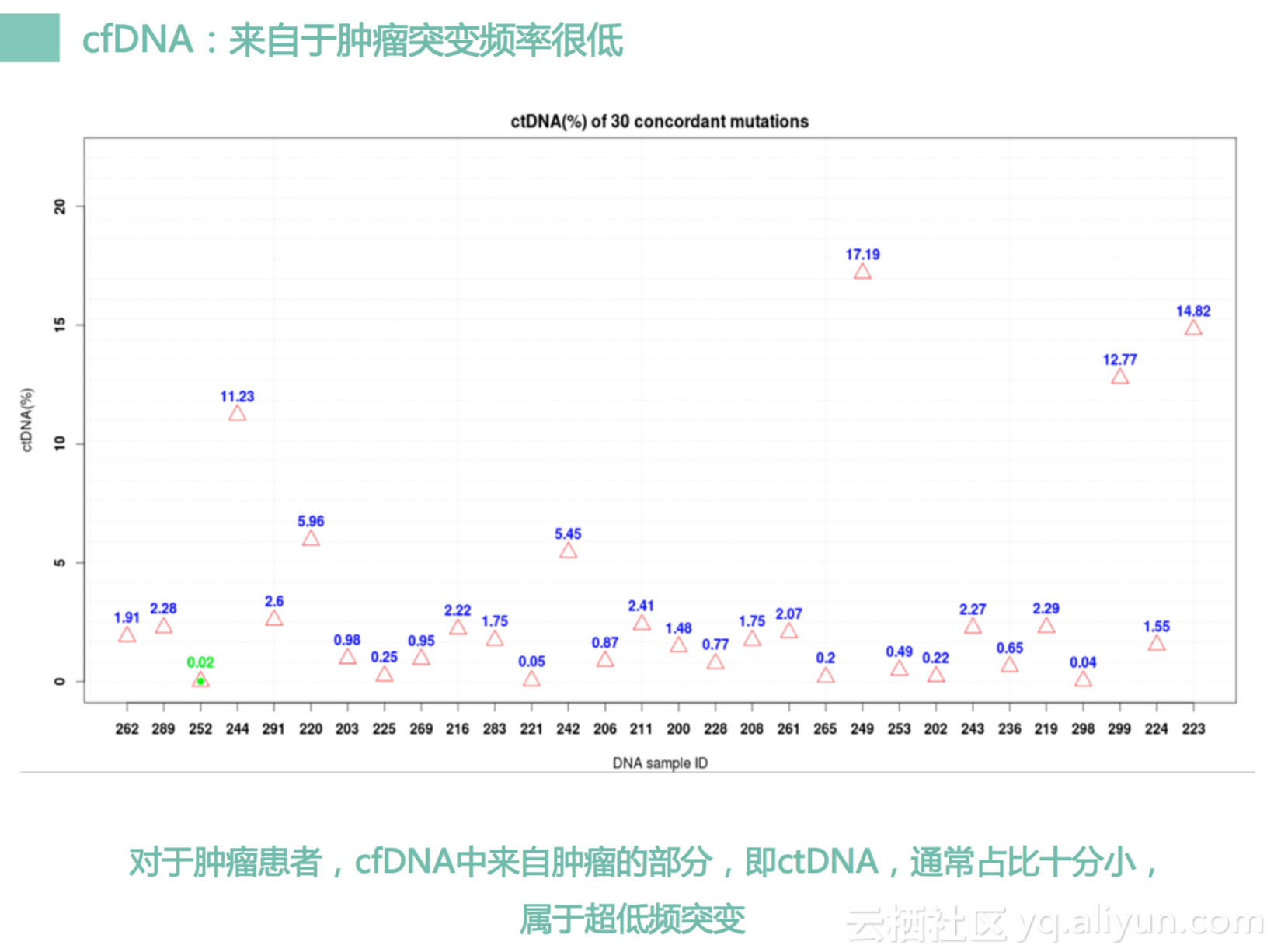
所以这个cfDNA测序分析是一个很好的东西,但也是是一个很难的事情。因为来自于我们肿瘤部分的突变总是很少,我们健康人的身体内部也会有大量的cfDNA,而且我们也测过很多健康的cfDNA它的量并不会比肿瘤病人少多少,大部分时候是持平的,只有一些晚期的病人才会很多很多,所以健康人跟早期患者其实基本上没有差别。然后癌症患者的cfDNA里面,它也会有大量的DNA是来自健康的细胞,只有少部分来自肿瘤细胞,可能不超过5%,一般时候是在1%上下,有时候甚至是千分之一左右的量。那我们要捕获的是千分之一的DNA变异。要想测出这千分之一水平的DNA变异,我们就需要测序测得很深才行。
然后另外一个问题是,我们健康人也会有基因突变,这个很好奇吧。就是一般人即便一个很健康的人也会有基因突变,突变的数量也不会特别特别少,其实也蛮多的。它只是因为它突变的位置不是那么重要,并不会引起癌症。但是突变因为是一种细胞的自然状态,它是一定会发生,只要细胞不发生分裂,会有受损伤,它就一定会突变,特别是一些吸烟的人群。他的突变率会比正常人还高一些。所以我们不能够光通过突变就认为他是一个癌症患者,我们需要通过很多方法把它区分出来。所以这一块有很多技术优化的方法。
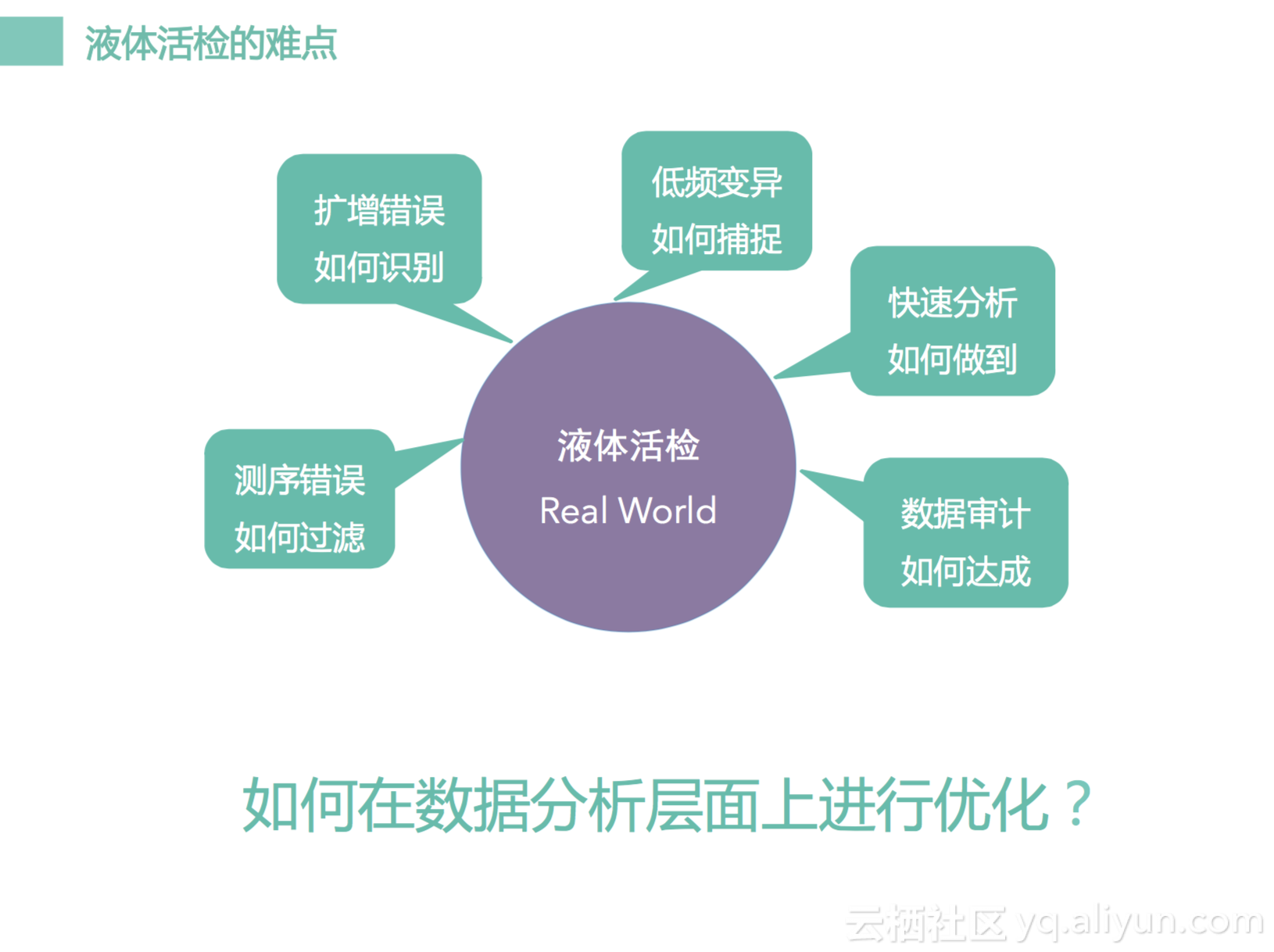
所以这一块现在有很多问题,第一个是突变频率很低,有时候是千分之一,百分之一这样水平,那我们怎么样区别它。另一个是测率错误。因为我们知道任何信号采样都伴随有噪声,因为我们测序也是一种采样过程,任何信号都伴有噪声,那噪声来自与哪里呢?来自于测序的错误,来自于扩增的错误,来自一些低频的突变可能是一种假阳性,那怎么样去区别它,这是一个真阳性还是假阳性,它是一个真实的信号还是一个噪声。因为突变频率很低,只有千分之一,百分之一这样的频率,我们怎么样才能够不漏掉它,怎么样才能做到阳性符合率很高,不会漏掉很多信息,不会有太多假阳性出来。那我们需要做很多很多测序的方法去优化它,然后怎么去做快速分析。因为我们海普洛斯做的是临床服务,临床服务讲究两个点,一个是快一个是稳,怎么样又快又稳把我们服务去传递给我们的患者,给我们医生是一个很重要的问题。然后因为我们做临床服务,所以做的事情是很严肃的事情。我们怎么样通过一些方法去做数字审计去评判我们结果里面有没有问题,有没有可能是有纰漏的地方。我们先看看几个方法,就是我们怎么样去排除一些错误呢?
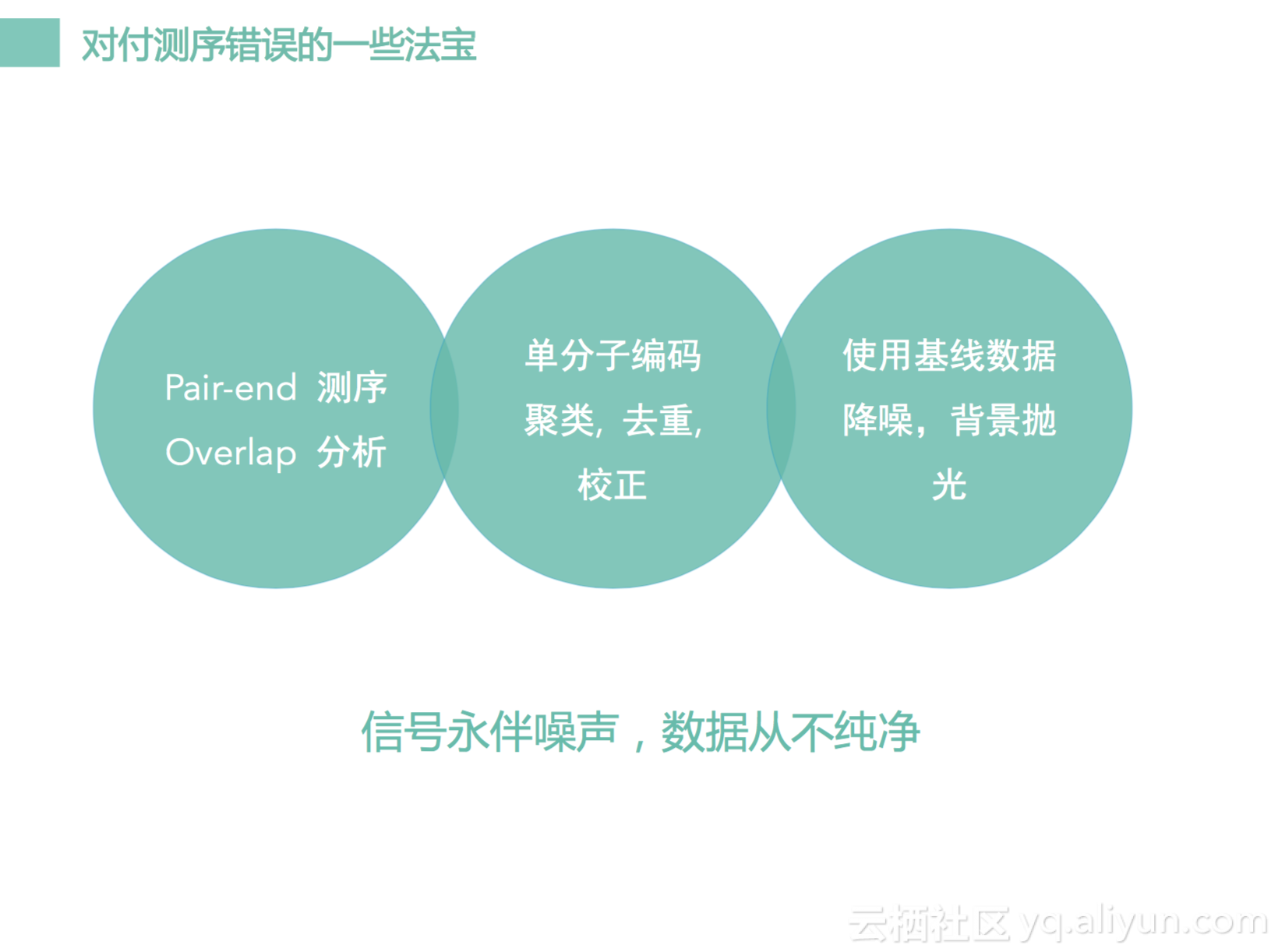
有几个方法。第一个我们现在测序大部分都是pair-end测序,就是同一个DNA模板会从两端测过来。两端测过来之后它会有一个地方是overlap的,可以做一致性分析。另外我们可以做单分子编码,这个技术在这一两年是十分十分火的技术,因为所有做肿瘤的技术里面,这个是十分重要的。那之前都没有开发出来,只有在这一两年变的十分火热。第三个我们可以用基线数据,我们可以基于基线做降噪,可以做背景抛光。一定要知道信号永远帮噪声,数据从来不会干净。我们一定不要去期望获得一个很干净的数据,我们一定获得充满了噪声的数据,怎么样通过噪声数据里面去发现一些真实有价值的突变。这是我们的一些工作。
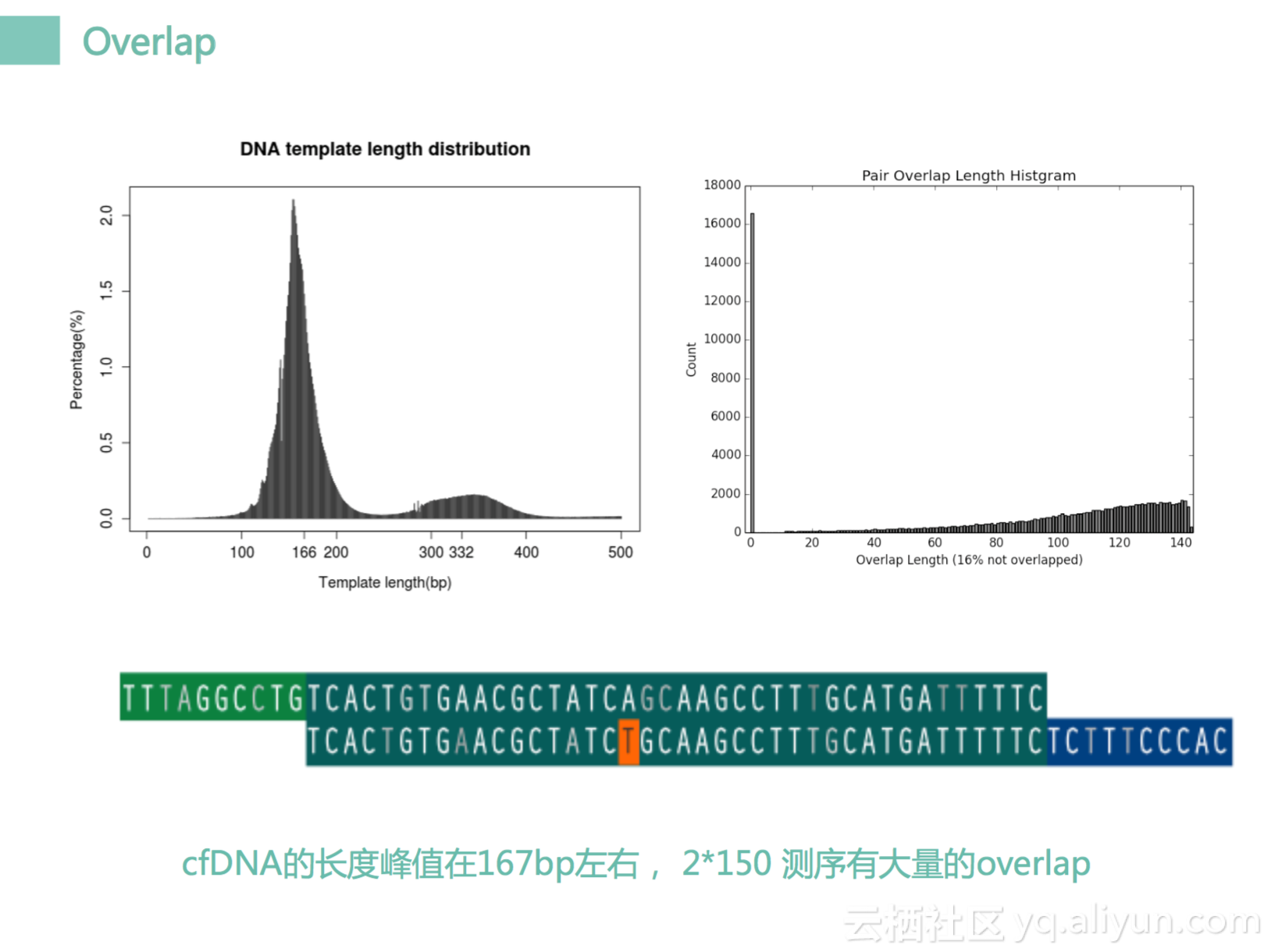
先看第一个overlap分析方法,我们看到左边这个图其实来自于我们一个分析图。这个是我们cfDNA,就是我们体内循环DNA的一个长度,它的长度是有规律的,是有峰值的,第一个峰值是在166BP左右,就是166个碱基,然后这个大小刚好是我们细胞里边核小体的一圈,所以它是一圈一圈掉落的,是有规律的。然后第二个的峰值就是一个小峰刚好是在332左右,其实刚好是两圈。所以我们知道循环DNA里边长度大小大部分是一圈166bp左右,以及一部分两圈,以一圈为主。然后我们测序的话,一般是测什么,2×150的测序。我不知道在座各位有多少是搞测序的,2×150的测序的话,我们知道166BP的长度DNA你这边测过来,这边是150,其实它会有110,120它是重复的。120重复数据我们是测过两片的,你看这一段DNA,它是测过两遍的。那测过两遍的话,overlap区域中每一个碱基的信号我们经过了一个双重确认的一个过程。如果它们100%吻合了,其实他是一个很好的情况,但是总是会有一些地方,它会两边都对不上,就是一个mismatch。我们看这里有一个T,它是对不上的。上面是A,下面是T。那怎么办呢?一种方式我们把两个点都去掉,另外一个方式我们把它进行纠正。如果这个A的质量很高,T的质量真的很低的话,我们会认为T是一个错误,把T进行一个校正,所以这样子的话,我们可以获得大量的错误校正,而且这个核校正可以获得一个统计图。这个统计图后面会指导我们做好几个事情。
然后我们开发了一个工具,这个工具我们是开源的,叫AfterQC,它可以全自动化地进行校正,而且可以把所有信息进行统计。这个工具是自动化,批量化的,它可以同时处理几百个文件也可以。

同时这个软件可以去接头,什么意思呢?如果我们的DNA长度没有到150,比如说140长度。那它测完140之后,后面一段我们称之为是adapter,就是说原始DNA它会连接一个adapter,而adapter它会被测出来。所以通过我们序列overlap的分析,我们会发现这个DNA原来比我们150更短。那我们会把另外adapter砍掉。所以我们可以做一个方法,就是可以不需要利用任何的adapter的序列信息,就可以把adapter取出来,把它砍掉。因为adapter污染会带来很多假阳性错误,如果不分析的话,它是会产生假阳性的,那我们基于这个方法会它砍掉。

另外有意思的事情,可能大部分人都没有注意到,我们测序仪里面会产生很多的气泡,就是我们测序仪里面是一个泳道一个泳道,它可能会鼓起一个气泡来。这个气泡的话,它会产生一些假信号,特别是会产生一些连续的信号。比如说都是G,polyG,是因为它的气泡产生之后,它会阻挡我们测序周期里面的清洗过程,就是鼓一个泡出来之后,它不能被洗掉了,就会产生一个连续的信号。所以我们可以通过这个连续信号可以去识别他,里边是不是有一个气泡。那我们看这个图,这其实是一个Illumina NextSeq500,然后这个地方是每一个图片,我们发现这里面有一个气泡把它鼓起来,这里面是polyG。
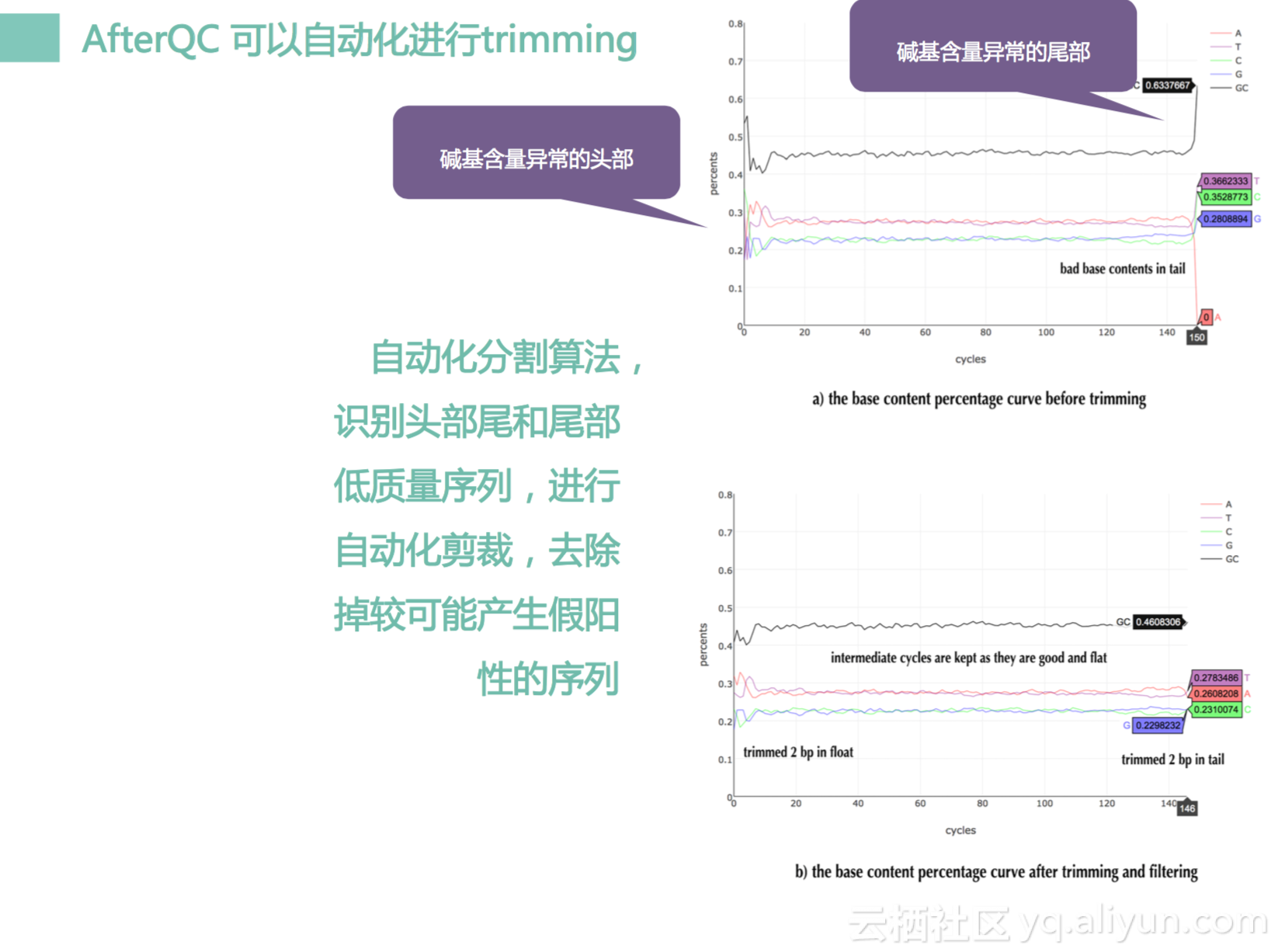
然后AfterQC还可以做自动化的剪裁,什么意思呢?比如我们测序总共有150多的cycle,前面几个,后面几个其实会比较容易出错误,波动会比较大。如果不把它去掉的话,它会对我们数据产生影响,那对于一些常规分析,其实没有关系。因为它不太在意这样一些低频突变,但是对于我们肿瘤分析,他是很关键的。为什么呢?因为你如果不它去掉,它就会产生一些假的变异。假变异会让我们认为是一个突变,我们会去做后续分析,如果我们产生假突变之后我们没有把它去掉,我们会给出一个临床当中一个假反馈。
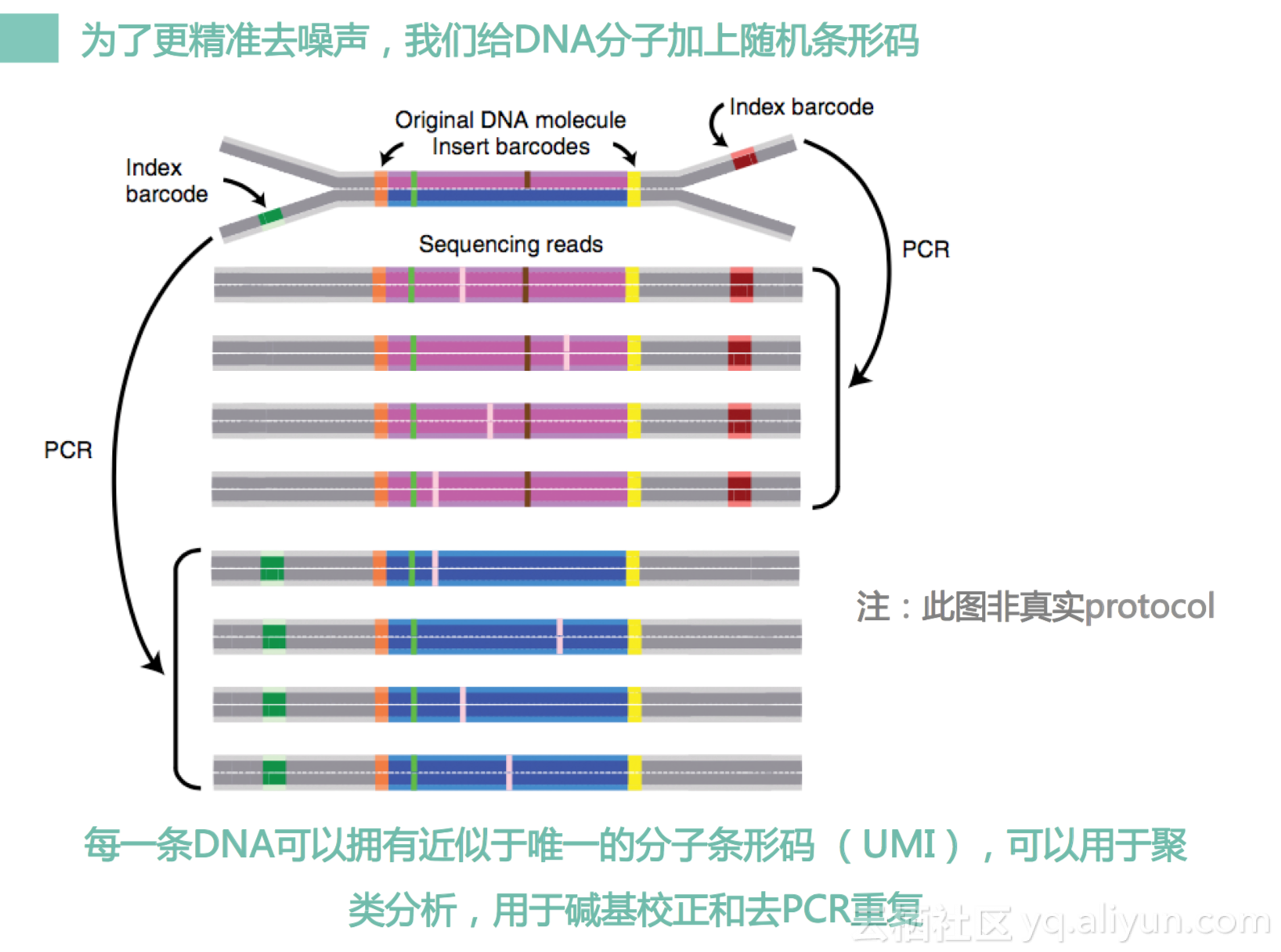
我们还开发了单分子编码方法,基于我们的方法把很多相同模板来源的DNA序列进行聚类。

比如说这个是三条DNA,三条DNA是来自于同一个原始DNA,是经过我们PCR重复出来的,然后它会有一些位点是不一样的。比如说下面两个地方是C,这个是G,这个地方上面下面是T,然后这个有一个删除,我们可以通过这个方法,我们把这个比较错误的位置把它找出来,找出来之后把它去掉。去掉之后,我们可以获得一个很干净的结果。

比如说这样子一个图,这个是我们的一个测序结果。如果我们不使用我们的deemi方法过滤的话,它是一个很脏的,有很多错误的数据,看起来像是满满的突变。

如果我们用我们方法过滤的话,它是一个很干净的图,它里面已经没有什么噪声,所以我们可以通过我们的方法把很多很多错误给去掉,把这个噪声降的很低。
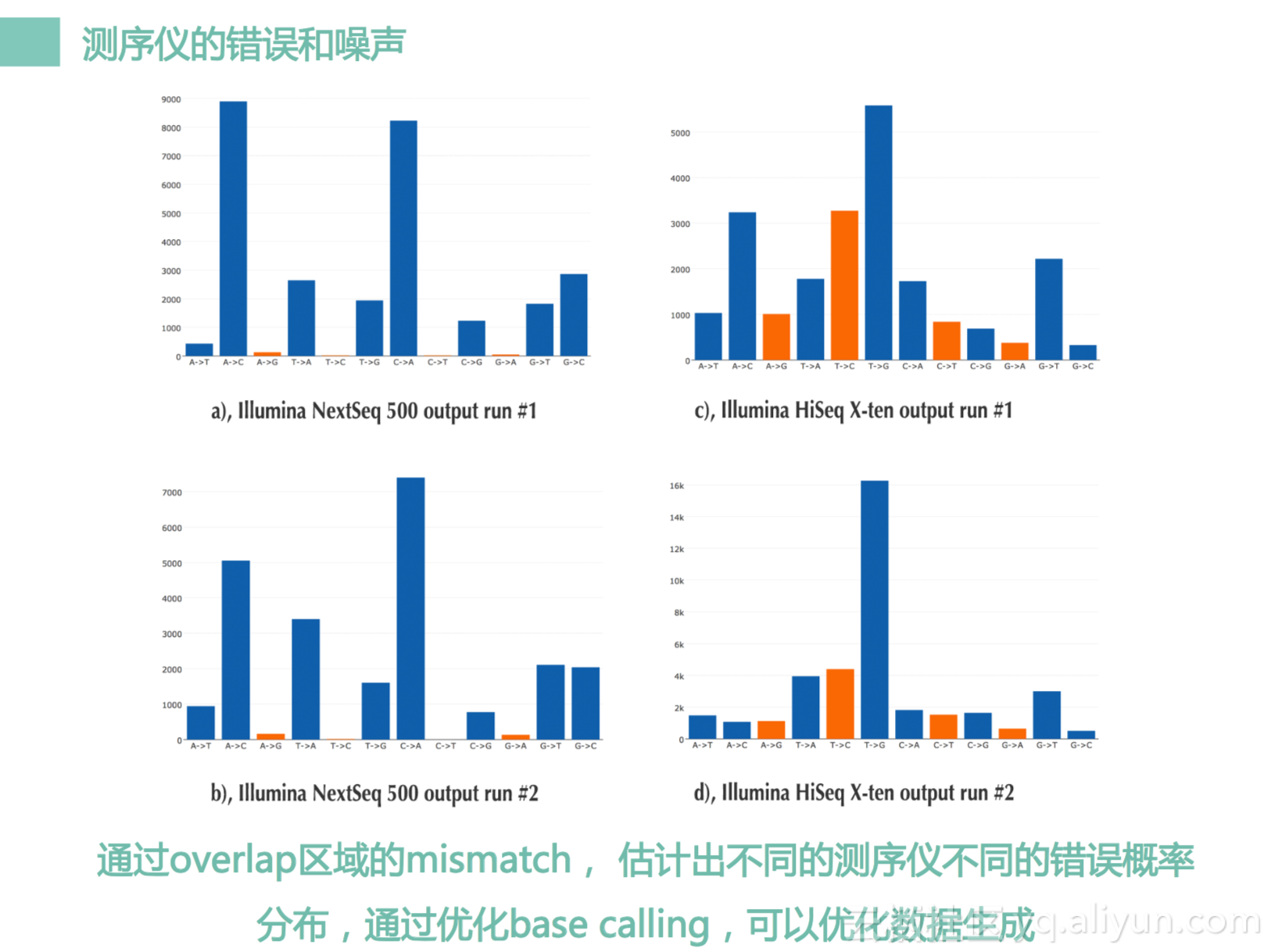
然后我们又发现,其实不同的测序仪,其实是有不同测序错误的pattern的我们看看这个图。左边两个图来自于NextSeq 500,右边两个图来自于HiSeq x10。我们看到左边这个图,这下面下面是A到G,是G到A,这个是C到T,T到C,会发现NextSeq中,A到G,是G到A,C到T,T到C的图片会很少。为什么呢?因为NextSeq它的原理是一个两色发光法,就是红光是C,绿光是T,A是两个都发光,G是两个都不发光。所以你看我们A,A是两个都发光,G是两个都不发光,所以我们很少会碰到两个都发光的信号,会被误认为两个都不发光。就是说两个都发光的信号跟两个都不发光的信号,其实差别是很大,它是可以通过我们程序很好把它识别出来的。而我们T和C,一个是发了红光,一个发了绿光,因为他们信号强弱其实不在意,但是它们频率是不一样的,频率一般是不会搞错的,所以T到C之间错误也跟少。这边是一个HiSeq,四色发光原理,ATCG都有不同频率的光代表,所以它们错误频谱会比较均匀一些。但它也有一些相似性,就跟这边NextSeq不太一样,这边就很奇葩。所以我们通过这个方法,我们可以去修改我们base calling,改完之后的话,我们可以去抑制比如说A到T之间的错误,抑制完之后让A到T之间往上面走一点点,把这四个做的更平缓一点点。所以的话,我们可以降低整体的错误率,让我们的噪声降低一些。
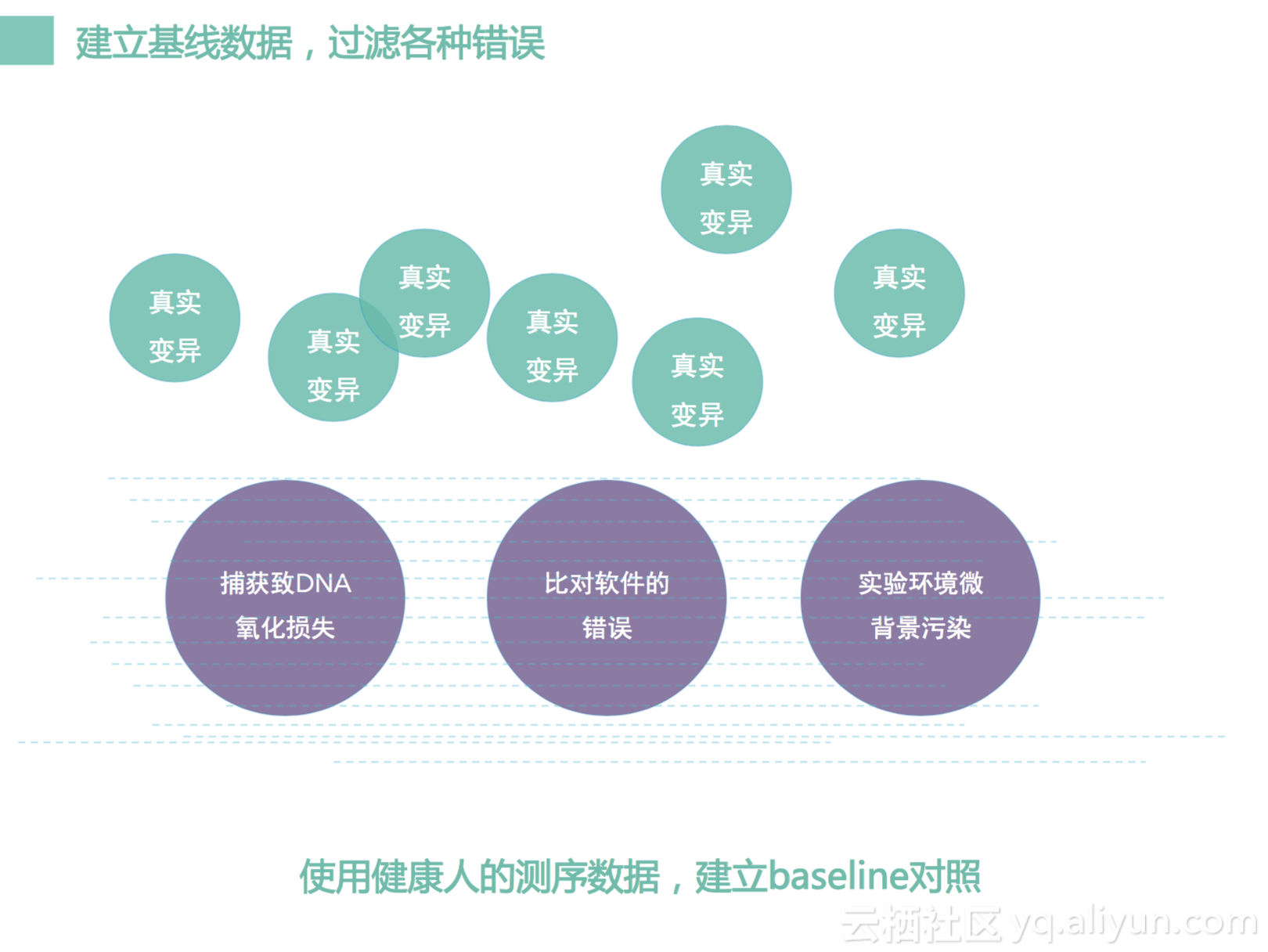
另外,我们做了一个背景基线分析。我们PCR扩增,以及很多的实验过程,比如说捕获,都会产生很多的错误。这个各种各样的错误,很多时候不能够光靠一个数据特征去把它去除。我们把很多批次,很多不同人数据放到一起来,做成一个基线,可以看作是图里的水面。那错误会填满这个水面基线,真正的变异的话,它一定要高于我们的基线,即我们信号的强弱要高于我们的基线,我们才认为它是一个真阳性信号,它如果是在基线下面,则可能是一个假的阳性。所以通过我们的基线我们可以去识别出来里边的变异到底是一个真阳性还是假阳性。
所以我们做了很多很多事情去过滤去降低这个背景,但是有的时候会适得其反,我们每一步都过滤很多的话,我们就会发现很多很多变异我们call不出来了,为什么呢?我们在前面一步两步三步四步我们都做了过滤,导致有一些数据,可能是一个真阳性会被我们去掉,对不对。那如果被我们去掉的是临床里面一个很重要的变异,这是不可以接受的。

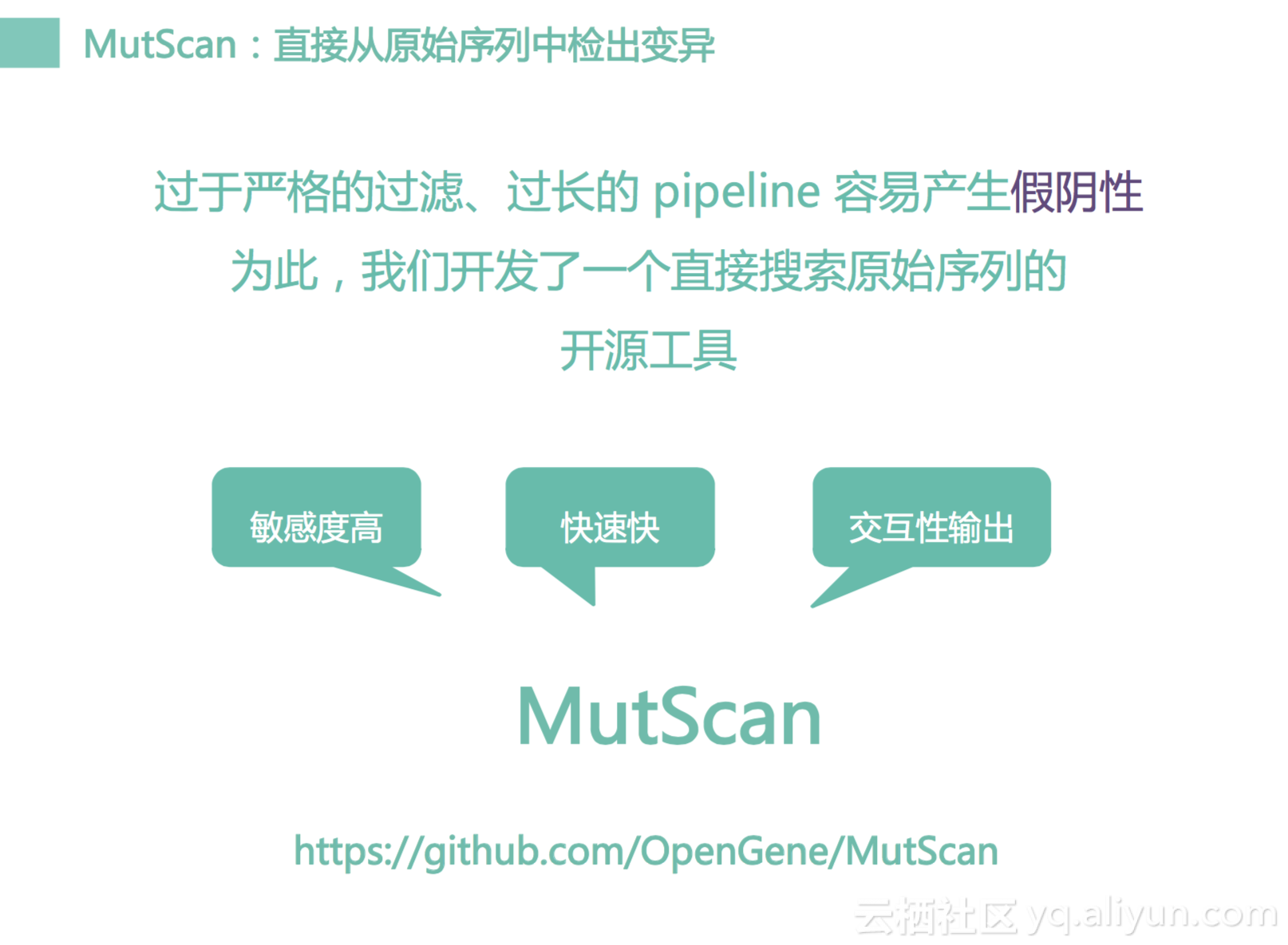
那怎么办呢?我们开发了另外一个工具,我们称之为MutScan,这个工具可以从头到尾不需要经过任何的pipeline,直接去搜索我们FastQ文件,搜索完之后就可以给出这个样本,它到底有没有这些重要变异,它是很快速的,具有很高敏感度,而且可以有HTML的输出。这是一个图,我们可以以50倍的速度去搜索原始序列,然后对于每一个变异我们可以输出一个堆叠图,这个图很像是一个pileup,会把每一个序列往上面堆,进行对齐里面里面的变异值都会输出来。这样是一个很直观很快速的方法,基于这个方法的话,我们可以去排出假阳性的错误。
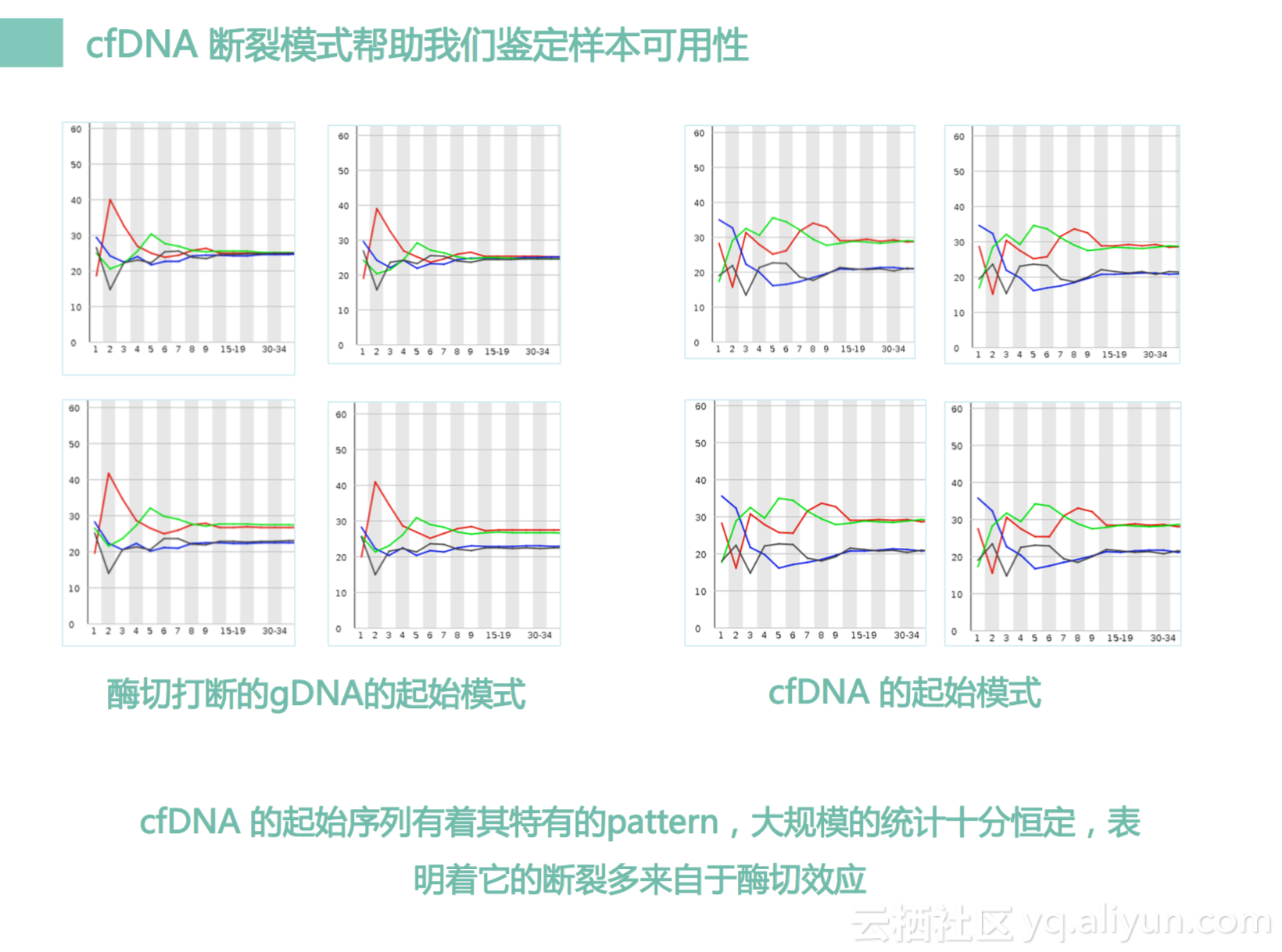
然后我刚才说过我们cfDNA是有一些特异性的。而且它不但长度有特异性,其实它的头部pattern也有特异性。什么意思呢?我们这个地方截取了我们前面十个碱基的图,左边是我们gDNA,就是我们白细胞里面DNA拿出来打断的,右边是cfDNA,cfDNA比较短,所以没有经过打断。这个gDNA是我们用酶把它打断的,所以它是有起始pattern的,它们的图是很像很像的。但是cfDNA这个图也很像,但是它没有经过酶打断就那么相似,表示什么,表示我们cfDNA本身就有一个pattern,在我们细胞内部是怎么转的呢,可能也是有一些酶的偏好性,导致这个pattern产生。那基于这个pattern我们可以做一些事情,什么事情呢?我们可以去判断一个样本,到底是不是来自于cfDNA,还是gDNA还是来自FFPE还是来自尿液,每一种DNA都会有一种pattern。基于这种pattern数据,我们大概统计了超过5千个样本的数据。

我们建立了一个机器学习模型,这个模型可以达到大概只有千分之一错误率,去判断一个样本,到底是来自于cfDNA还是非cfDNA,这个有什么价值呢?这个价值有两点,第一点我们在实验室里面有时候很多时候是会发生的错误的,特别是人会发生错误,人有时候可能会错误把一个样本当成另外一个样本。因为我们测序会测两种,一种是gDNA,一种是cfDNA。如果实验员犯错误,会有一定的机率搞反掉,搞反掉如果我们分析的时候也搞反了,那我们会产生一个错误的结果。但是现在,我们的软件可以把它识别出来,杜绝这种情况的发生。
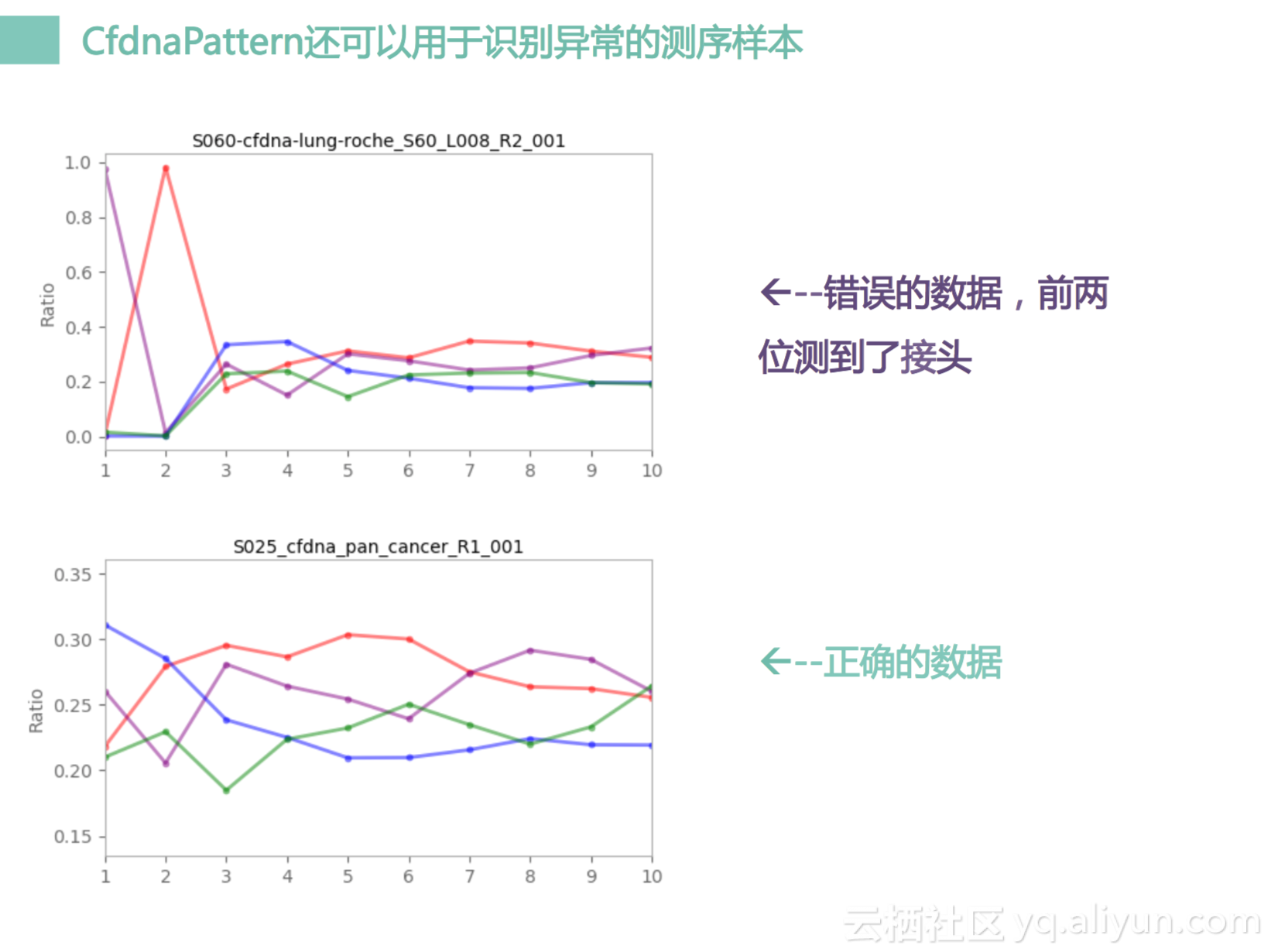
另外一个就是我们在测序的时候会有很多异常,比如说上面这个图,它有两个是很怪的,到了100%的比例,什么意思呢?其实是它测了接头,这个接头测完之后会产生一个比较奇怪的pattern,那么这个pattern会通不过我们的测试,然后被报错,报完错之后我们知道这是一个错误的信息我们要到实验室里面去检查,去回溯为什么这个会发生。所以在我们提供临床服务的时候,要对每一个细节都很仔细,很稳健,这样才会有一个高质量的服务。

然后我们说有很多很多的算法,其实在开发和测试的时候,我们还没有拿到真正的实验数据,这时候怎么办呢?比如说我们做单分子编码,我们的实验还没有完成,但是我们不能够等到实验完成了,再去开发软件,因为那样就都晚了,所以我们需要生成一些模拟数据。因为只有使用模拟的方法,才可以产生我们任意的,在理论上可以使用的数据。所以我们这边也开发了一个软件是SeqMaker,可以生成一些只要你需要的数据。比如说他可以是一个全基因组,也可以是一个捕获测序,深度可以很深到5万层,也可以是5千层,也可以是低深度3,4层,可以带有很多很多突变,也可以没有突变,也可以单倍体,可以双倍体。我们开发的这个软件,可以去模拟很多很多数据,可以产生很多变异。
同时更重要的事情是,它可以模拟很多的噪声,以及很多类型的错误,这些错误其实是在真实地仿真我们测序的过程。所以我们获得的数据不是那种很干净的数据,也是带有错误的数据,但是刚好可以考虑我们算法里面有没有能力可以去处理这个数据。所以这个是仿真性很强一个软件。
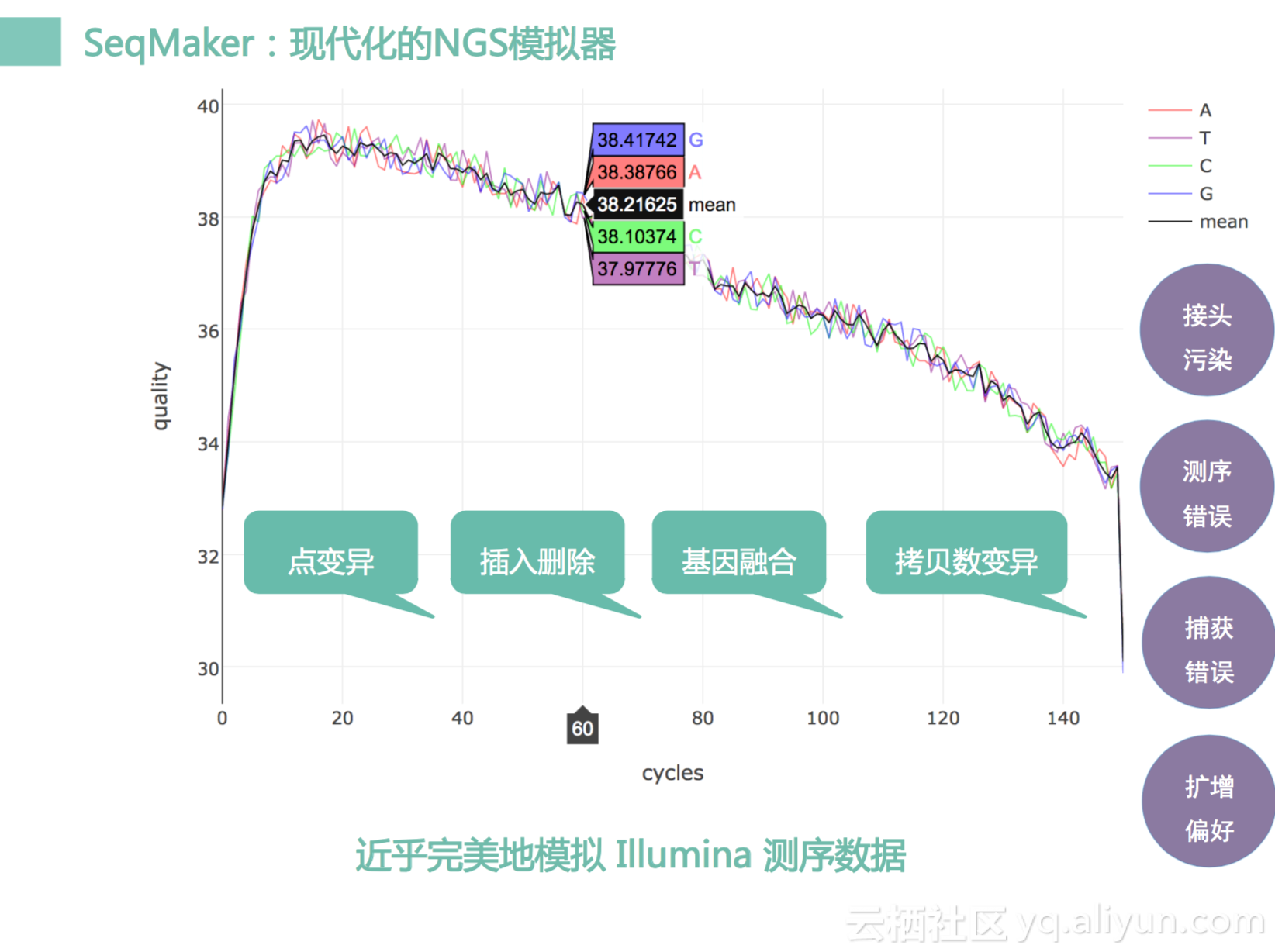
这个图是我们通过仿真之后的数据,看到这个quality曲线,这个我们看它有开始的质量有一个上升的过程,然后开始逐渐下降,到最后一个base质量是十分低的,其实这个过程其实很像我们测序过程。同时它会模拟我们接头污染,捕获错误等等。同时这个软件可以产生一些我们可配置的点变异,INDEL,基因融合,拷贝数变异等。
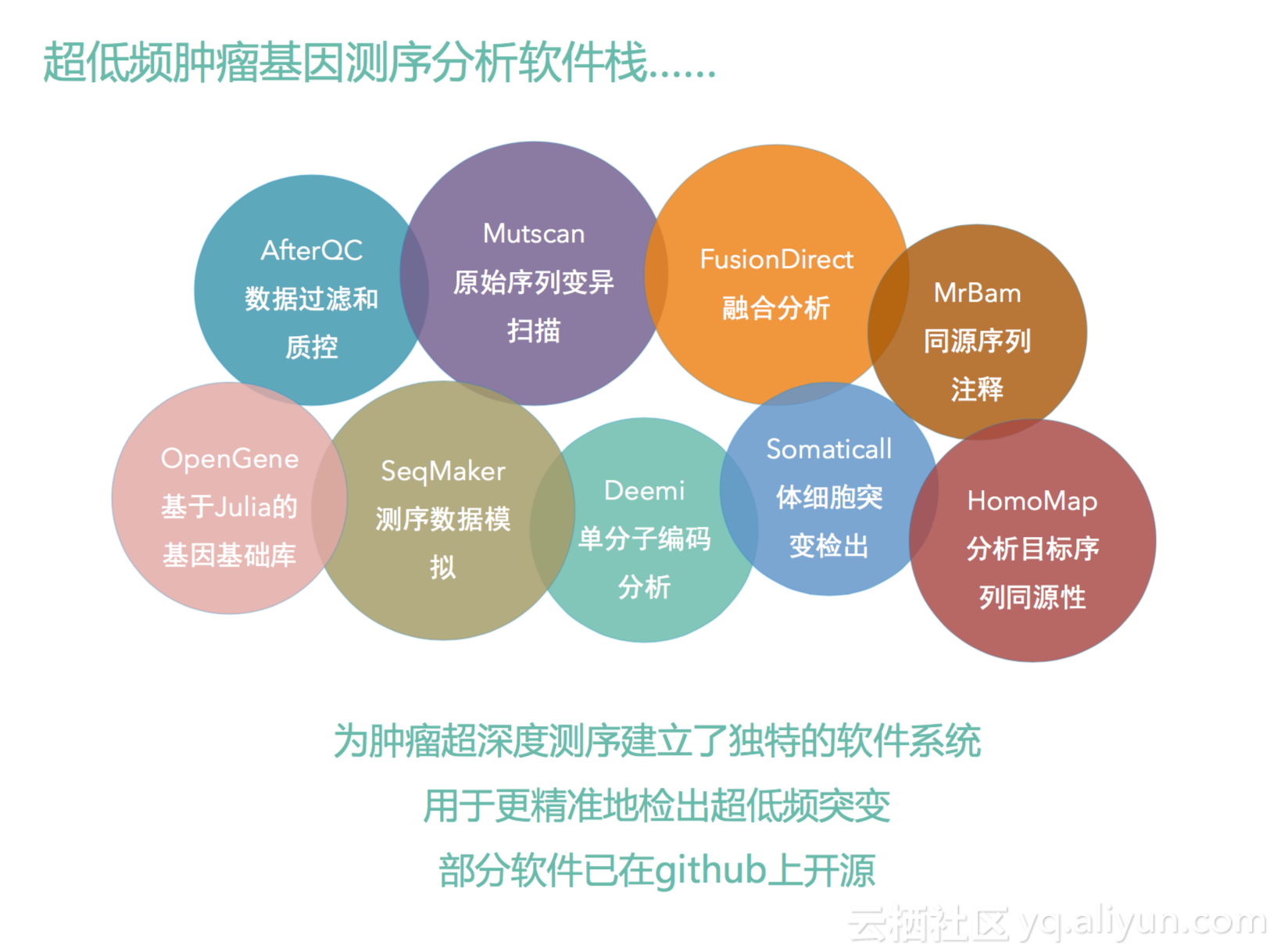
我们开发了一系列的软件栈,这里面包括了刚才说的这些软件,这里我就不一一讲了,因为时间有限。我们做这些事情的原因就是因为当前的软件,就是比如我说GATK,它其实不是为了我们做肿瘤基因分析的用的,其实是为了做一些遗传突变分析用的,但是对我们做肿瘤检测,对我们超低频,超深度测序,它是没有办法很好地使用的。所以我们从头到尾重做了这样一系列工具,让我们可以更高敏感度,更好地分析肿瘤基因变异。


所以我们把知识把数据把样本把临床信息同时进行整合,整合完之后我们就可以进行大数据的挖掘,因为我们的目的不是单纯地做这个个性化用药,我们是希望我们有一天可以基于我们的数据,基于我们的方法,可以去做这个癌症早期的筛查。这个筛查我们需要利用基因大数据为依托,使用机器学习为手段,通过我们这个液体活检的方法做主要途径,进行肿瘤的最早期的筛查,可能是在一期,甚至更早的时候筛查。在这些方面,我们已进行了一些尝试。
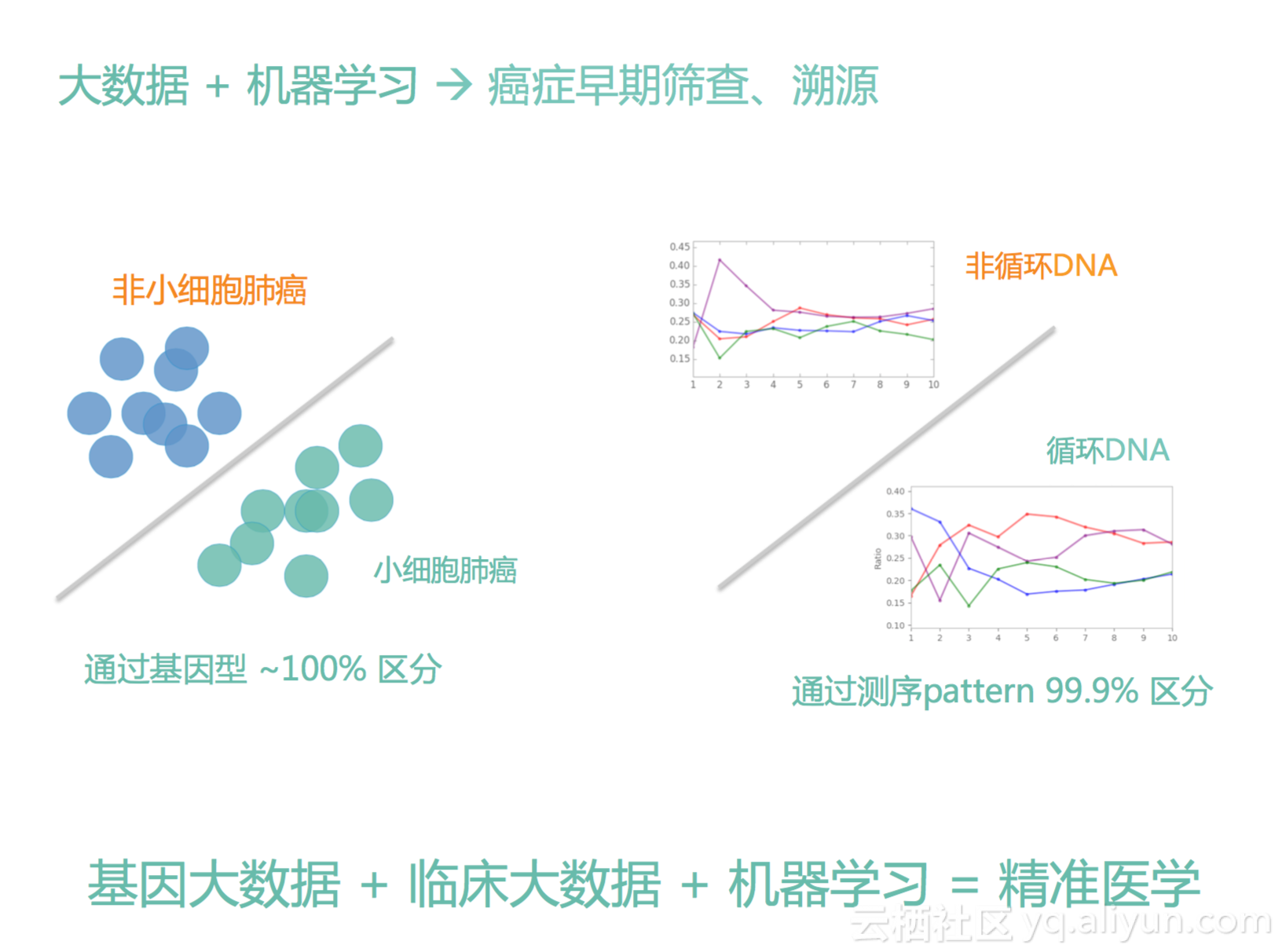
第一个我们仅使用基因型,我们可以把小细胞肺癌跟非小细胞肺癌百分之百分开,我们知道,就是说同是肺癌,不同的亚型,其实是有很多差别的,小细胞肺癌它其实一般来说不会有什么EGFR突变,非小细胞肺癌较大可能会有EGFR突变,基于我们的方法可以仅通过基因型给百分之百区分开来,然后我刚才说过,我们通过cfDNA DNA的pattern,我们可以做很好很好的一个区分,区分它到底是不是来自cfDNA还是来自于非cfDNA,我们的结果可以到千分之一的错误率左右,这已经是一个很高的分辨率。我认为,组学大数据+临床大数据+机器学习等于精准医学,所以以后,我们不能够光看一个点,我们一定是结合起来看,而且我们的方法一定要往前看,一定要结合人工智能的方法才可以真正的让我们的分析更精准。
做这个大数据的话,光靠一家机构单打独斗肯定是不够的,只有靠我们所有的机构在一起,我们把数据进行共享进行统一进行存储,进行挖掘之后才是真正的一个大数据,所以我们一定会去推动去参与整个行业里面去建立一个癌症基因大数据的一个共享。但这个事情会很难做,为什么呢?因为每一个机构,每一个企业都会有自己的想法,会想如果我把它分享出来之后我会有什么收获,我会有什么风险,但是一定会有一天,我们中国人,特别是我们中国人,会把我们这个所有的肿瘤数据放到一块儿来,形成一个中国人的肿瘤基因大数据,这样我们可以基于这个数据库可以做更好的挖掘,可以统一中国人的突变频谱,基于中国人的数据去做制药,做后面的临床实验。直到有一天我们真的可以说,我们只要发现某一个变异,我们就可以吃什么药。这确实会有一点困难,但是我们会希望跟一些公司,在小范围之内先去进行尝试数据共享,基于共享的数据更好地服务患者,最终实现我们公司的愿景,即让每一个生命健康120年。