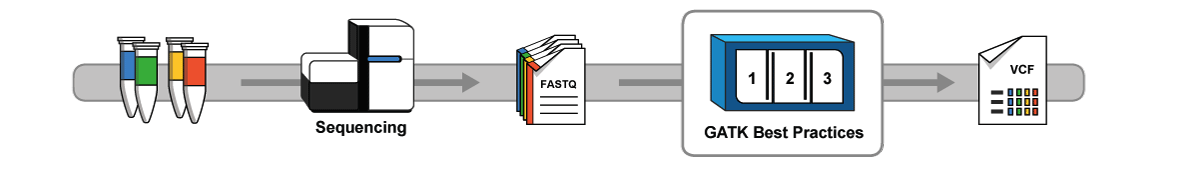
GATK,全称是Genome Anlysis Toolkit,顾名思义,是一套用于分析基因组的工具箱。主要功能是寻找变异位点和基因分型,但是实际上功能超多,导致初学者都不知道从何学习GATK。
为了帮助听说过GATK的人更好的入门GATK, 我,作为一个初学者,在这里介绍一下我的经验。
虽然GATK的功能超级多,但是主要可以归为以下几个方面:
- 诊断和质量控制工具(Diagnostics and Quality Control Tools)
- 序列数据处理工具(Sequence Data Processing Tools)
- 变异位点探索工具(Variant Discovery Tools)
- 变异位点评估工具(Variant Evaluation Tools)
- 变异位点操作工具(Variant Manipulation Tools)
- 注释模块
- 读段(reads)过滤
- 资源文件解码工具
- 参考序列实用工具
如何快速建立GATK的心理表征
然后这里面的每一项点开都有好多内容,我第一次点开的时候,也是一脸茫然,不知道从何入手。
但是根据《认知学习法》,最好的学习方式就是“不要怂,直接上”,找到一个已有流程,先把代码敲上去,然后慢慢理解每一行代码的作用,建立一个模糊的心理表征,然后循序渐进,慢慢学习其他工具,最后就能熟练使用GATK了。
所以记下来主要的任务,就是带大家建立关于GATK的模糊概念。
mapping-by-sequencing其中一个重要环节就是“SNP calling”,我最初用的是samtools和bcftools,结果的variant特别多(估计很多是假阳性).虽然最后还是找到了causual mutation, 但是为了保证今后causual mutation的准确性,我发现了有文章使用了GATK。他给的代码如下:
1. Add read groups (Picard tools)
AddOrReplaceReadGroups.jar I=sorted.bam_file O=s1.rg.bam RGLB=genome RGPL=ILLUMINA
RGPU=GATKv4 RGSM=sample_name VALIDATION_STRINGENCY=LENIENT
2. Mark duplicates (Picard tools)
MarkDuplicates.jar INPUT=s1.rg.bam OUTPUT=s2.dedup.bam ASSUME_SORTED=TRUE
VALIDATION_STRINGENCY=LENIENT METRICS_FILE=s2.dedup.metrics
3. Index (samtools)
samtools index s2.dedup.bam
4. Realign reads (create intervals first, then do IndelRealigner) (GATK)
GenomeAnalysisTK.jar -I s2.dedup.bam -R ref_file -T RealignerTargetCreator -o s3.intervals
GenomeAnalysisTK.jar -T IndelRealigner -I s2.dedup.bam -R ref_file -targetIntervals s3.intervals -o
s4.realn.bam
5. Unified genotyper (GATK)
GenomeAnalysisTK.jar -T UnifiedGenotyper -R ref_file -I s4.realn.bam -glm BOTH -o s5.UG1.vcf -mbq 30 -nt
4
6. Base score recalibrator (GATK)
GenomeAnalysisTK.jar -T BaseRecalibrator -I s4.realn.bam -R ref_file -knownSites s5.UG1.vcf -o s6.recal
7. Print Reads (GATK)
GenomeAnalysisTK.jar -T PrintReads -R ref_file -I s4.realn.bam -BQSR s6.recal -o s7.recal.bam
8. Unified Genotype (GATK)
GenomeAnalysisTK.jar -T UnifiedGenotyper -R ref_file -I s7.recal.bam -glm BOTH -o s8.UG2.vcf -mbq 30
9. Base score recalibrator (GATK)
GenomeAnalysisTK.jar -T BaseRecalibrator -I s4.realn.bam -R ref_file -knownSites s8.UG1.vcf -o s9.recal
10. Print Reads (GATK)
GenomeAnalysisTK.jar -T PrintReads -R ref_file -I s4.realn.bam -BQSR s9.recal -o s10.recal.bam
11. Unified Genotyper (GATK)
GenomeAnalysisTK.jar -T UnifiedGenotyper -R ref_file -I s10.recal.bam -glm BOTH
第一步: 不管三七二十一直接把代码自己敲一遍。
第二步: 读每行代码的解释,理解他的意图
- 用了
picard tools的AddOrReplaceReadGroups.jar加了read group,根据以前百度GATK的经验,了解GATK要求bam文件的header必须包含@RG,所以这一步应该是前面比对时候,没有在参数中增加相应部分。所以如果我在比对的时候增加了这个参数,这一步就可以免了。 - 用
picard tools的MarkDuplicates.jar去重。这是因为二代测序有一部桥式PCR扩增底物的过程,在100x以上出现reads重复,很大可能是PCR扩增重复了,所以可以直接去掉。 -
samtools的index,建立索引,提高检索效率。 - 先建立indel区间文件,然后对该区域进行reads重比对。这一步我没有经验,然后我在GATK的论坛搜索了这个工具,发现原因是indel会导致附近的错配,所以需要借由这一步降低indel附近的假阳性。但是最新的
HaplotypeCaller or MuTect2已经不需要了,这些工具的haplotype组装步奏效果类似。这里的UnifiedGenotyper or the original MuTect.还是要的。 - 使用
UnifiedGenotyper寻找variant。论坛搜索发现,现在推荐用HaplotypeCaller,效果更好。 - 根据上一步得到vcf文件对第四步得到BAM文件进行碱基重校准,得到校准所需的文件。
- 使用
PrintReads根据第6步得到的文件,对第四步得到的BAM文件进行校准 - 根据重校准的BAM文件重新找variant。
- 根据第8步的文件进行校准
- 输出第二次校准后BAM文件
- 第三次寻找variant。
第三步:根据上述过程对拟南芥中使用GATK寻找variant建立大致的认识:
- 常规步骤: 先比对,比对后把SAM转成BAM然后排序,建立索引,去除PCR重复
- 对indel附近进行重新比对
- 由于拟南芥没有已知的VCF文件让UnifiedGenotyper学习,所以需要通两轮碱基校准和variant calling降低假阳性
第四步: 再看看别的流程,不断修改最初的认识。我通过翻文献又找到了一个perl脚本
# 第一行,UnifiedGenotyper
# 输出read-$i.snv-gatk-snp.vcf
&execsys("java -Xmx2G -jar $cfg{gatk_dir}/GenomeAnalysisTK.jar -T UnifiedGenotyper -R $cfg{genome} -I read-$i.recal.bam -D known_snp.conv -A AlleleBalance -stand_call_conf 50.0 -stand_emit_conf 10.0 -dcov 200 -glm SNP -out_mode EMIT_VARIANTS_ONLY -log log.UnifiedGenotyper-snp.$i -o read-$i.snv-gatk-snp.vcf");
# 第二行,VariantFiltration,
# 过滤read-$i.snv-gatk-snp.vcf得read-$i.snv-gatk-snp.fil.vcf
&execsys("java -Xmx2G -jar $cfg{gatk_dir}/GenomeAnalysisTK.jar -T VariantFiltration -R $cfg{genome} -V read-$i.snv-gatk-snp.vcf --clusterWindowSize 10 --filterExpression \"QUAL < 30.0 || QD < 5.0\" --filterName \"HARD_TO_VALIDATE\" -log log.VariantFiltration-snp.$i -o read-$i.snv-gatk-snp.fil.vcf");
# 第三行 UnifiedGenotyper
# 输出read-$i.snv-gatk-indel.vcf
&execsys("java -Xmx2G -jar $cfg{gatk_dir}/GenomeAnalysisTK.jar -T UnifiedGenotyper -R $cfg{genome} -I read-$i.recal.bam -D known_snp.conv -A AlleleBalance -stand_call_conf 50.0 -stand_emit_conf 10.0 -dcov 200 -glm INDEL -out_mode EMIT_VARIANTS_ONLY -log log.UnifiedGenotyper-indel.$i -o read-$i.snv-gatk-indel.vcf");
# 第四行 VariantFiltration
# 过滤read-$i.snv-gatk-indel.vcf得read-$i.snv-gatk-indel.fil.vcf
&execsys("java -Xmx2G -jar $cfg{gatk_dir}/GenomeAnalysisTK.jar -T VariantFiltration -R $cfg{genome} -V read-$i.snv-gatk-indel.vcf --clusterWindowSize 10 --filterExpression \"QUAL < 10.0\" --filterName \"HARD_TO_VALIDATE_1\" --filterExpression \"MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)\" --filterName \"HARD_TO_VALIDATE_2\" -log log.VariantFiltration-indel.$i -o read-$i.snv-gatk-indel.fil.vcf");
#第五行 CombineVariants
# 结合read-$i.snv-gatk-snp.fil.vcf和read-$i.snv-gatk-indel.fil.vcf
&execsys("java -Xmx2G -jar $cfg{gatk_dir}/GenomeAnalysisTK.jar -T CombineVariants -R $cfg{genome} --variant read-$i.snv-gatk-snp.fil.vcf --variant read-$i.snv-gatk-indel.fil.vcf -o read-$i.snv-gatk.vcf.tmp -log log.CombineVariants.$i");
好像和之前建立的模型不太一样呀,这里变成先找variant然后过滤了。
但是,这些流程所用的工具基本都来自于
- 序列数据处理工具(Sequence Data Processing Tools)
- 变异位点探索工具(Variant Discovery Tools)
- 变异位点评估工具(Variant Evaluation Tools)
其实都是先找到variant,然后通过各种方法降低假阳性。
小结
总结一下:
GATK常用套路就是先找variant,然后降低假阳性(例如BQSR, VQSR或直接过滤等)
有了这样一个大致印象后,我再去学习GATK Best Practices,就稍微有点思路了。
这些就是我这段日子学习GATK的感悟,不涉及到具体的操作。希望大家能够提供更多GATK相关的pipeline,让我能够不断更新自己的心理表征。
吐槽
本来我是想麻烦我同学让他向师兄师姐要下他们的GATK流程,便于我借鉴模仿学习。但是他说他们老板认为这是机密,不能随便透露。那么问题来了,假设你不把代码公开,那么别人无法重复出你的生物信息分析结果,那该怎么办?当然吐槽归吐槽,我也能理解,毕竟要是被商业公司拿走,就可以拿出去挣钱了。
不过我相信,别人只能模仿你的代码,却无法模仿你的思维模式,所以我很乐意把自己的代码分享出去,只有经得起检验的代码才是好代码。