基因功能的注释依赖于上一步的基因结构预测,根据预测结果从基因组上提取翻译后的 蛋白序列 和主流的数据库进行比对,完成功能注释。常用数据库一共有以几种:
- Nr:NCBI官方非冗余蛋白数据库,包括PDB, Swiss-Prot, PIR, PRF; 如果要用DNA序列,就是nt库
- Pfam: 蛋白结构域注释的分类系统
- Swiss-Prot: 高质量的蛋白数据库,蛋白序列得到实验的验证
- KEGG: 代谢通路注释数据库.
- GO: 基因本体论注释数据库
除了以上几个比较通用的数据库外,其实还有很多小众数据库,应该根据课题研究和背景进行选择。注意,数据库本身并不能进行注释,你只是通过序列相似性进行搜索,而返回的结果你称之为注释。因此数据库和搜索工具要进行区分,所以你需要单独下载数据库和搜索工具,或者是同时下载包含数据库和搜索工具的安装包。
注意,后续分析中一定要保证你的蛋白序列中不能有代表氨基酸字符以外的字符,比如说有些软件会把最后一个终止密码子翻译成"."或者"*"
BLASTP
这一部分用到的数据库都是用BLASTP进行检索,基本都是四步发:下载数据库,构建BLASTP索引,数据库检索,结果整理。其中结果整理需要根据BLASTP的输出格式调整。
Nr的NCBI收集的最全的蛋白序列数据库,但是无论是用NCBI的BLAST还是用速度比较快DIAMOND对nr进行搜索,其实都没有利用好物种本身的信息。因此在RefSeq上下载对应物种的蛋白序列, 用BLASTP进行注释即可。
# download
wget -4 -nd -np -r 1 -A *.faa.gz ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plant/
mkdir -p ~/db/RefSeq
zcat *.gz > ~/db/RefSeq/plant.protein.faa
# build index
~/opt/biosoft/ncbi-blast-2.7.1+/bin/makeblastdb -in plant.protein.faa -dbtype prot -parse_seqids -title RefSeq_plant -out plant
# search
~/opt/biosoft/ncbi-blast-2.7.1+/bin/blastp -query protein.fa -out RefSeq_plant_blastp.xml -db ~/db/RefSeq/uniprot_sprot.fasta -evalue 1e-5 -outfmt 5 -num_threads 50 &
Swiss-Prot里收集了目前可信度最高的蛋白序列,一共有55w条记录,数据量比较小,
# download
wget -4 -q ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
gzip -d uniprot_sprot.fasta.gz
# builid index
~/opt/biosoft/ncbi-blast-2.7.1+/bin/makeblastdb -in uniprot_sprot.fasta -dbtype prot -title swiss_prot -parse_seqids
# search
~/opt/biosoft/ncbi-blast-2.7.1+/bin/blastp -query protein.fa -out swiss_prot.xml -db ~/db/swiss_prot/uniprot_sprot.fasta -evalue 1e-5 -outfmt 5 -num_threads 50 &
关于结果整理,已经有很多人写了脚本,比如说我搜索BLAST XML CSV,就找到了https://github.com/Sunhh/NGS_data_processing/blob/master/annot_tools/blast_xml_parse.py, 所以就不过多介绍。
InterProScan
下面介绍的工具是InterProScan, 从它的9G的体量就可以感受它的强大之处,一次运行同时实现多个信息注释。
- InterPro注释
- Pfam数据库注释(可以通过hmmscan搜索pfam数据库完成)
- GO注释(可以基于NR和Pfam等数据库,然后BLAST2GO完成,)
- Reactome通路注释,不同于KEGG
命令如下
./interproscan-5.29-68.0/interproscan.sh -appl Pfam -f TSV -i sample.fa -cpu 50 -b sample -goterms -iprlookup -pa
-appl告诉软件要执行哪些数据分析,勾选的越多,分析速度越慢,Pfam就行。
KEGG
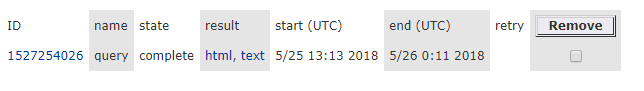
KEGG数据库目前本地版收费,在线版收费,所以只能将蛋白序列在KEGG服务器上运行。因此你需要在http://www.genome.jp/tools/kaas/选择合适的工具进行后续的分析。我上传的50M大小蛋白序列,在KEGG服务器上只需要运行8个小时,也就是晚上提交任务,白天回来干活。