提高自己分析能力的一个好的方法就是重复别人文章里的分析策略,所以这里会尝试对第一篇介绍R-ChIP技术文章"R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters"里的所有分析进行重复,我重复所用代码会更新在我的GitHub上,地址为https://github.com/xuzhougeng/R-ChIP-data-analysis
选择这篇文章进行重复的理由有三点:
- 一:最近要探索R-loop数据分析流程
- 二:这篇文章的通讯作者是大牛,Xiang-Dong Fu
- 三:这篇文章将分析所用代码都托管在https://github.com/Jia-Yu-Chen
背景知识
我整理下和数据分析有关的几个知识点:
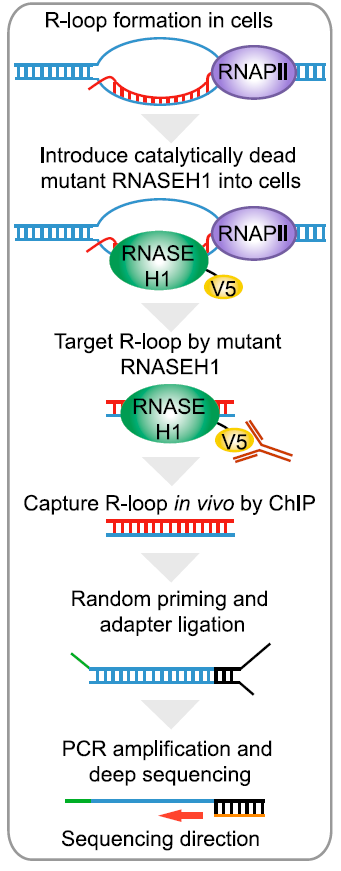
- R-loop是一种RNA/DNA三链结构体,与基因组稳定性和转录调控有关。
- 通过电镜观察,R-loop大小在150~500bp之间。
- 硫酸氢盐测序(bisulfate sequencing)表明R-loop主要出现在基因启动子的下游。
- R-loop所在非模板链(又称编码链)具有很强的序列偏好性,计算方式为(G-C)/(G+C)
R-loop的高通量分析方法目前都是依赖于S9.6抗体捕获RNA/DNA杂合体,然后超声打断或酶切,如果后续对DNA进行测序,那就是DRIP-seq(DNA:RNA immunoprecipitation [DRIP] sequencing),如果后续对RNA逆转成的cDNA继续测序,那就是 [DRIPc]-seq(DNA:RNA immunoprecipitation followed by cDNA conversion)。 然而酶切的分辨率不够,超声又容易破坏脆弱的R-loop结构,于是就导致目前很多文献报道有矛盾。
这篇文章就开发了一种新方法,基于RNase H的体内R-loop谱检测策略。作者构建一种没有催化活性,且在C端有一个V5标签的RNASE H1,RNASEH1与RNA/DNA结合,超声打碎,用anti-V5抗体进行染色体免疫共沉淀(ChIP)。随后RNA/DNA杂合体转换成双链DNA(ds-DNA), 之后便是链特异性测序。
关于链特异性测序,推荐拜读链特异性测序那点事
准备分析环境
软件部分
文章中"Software and Algorithms"这部分列出了分析主要所用的软件,加上下载SRA数据所需工具和一些常用软件,一共要安装的软件如下:
- SRA Toolkit: 数据下载工具
- Bowtie2: 比对工具
- SAMtools: SAM格式处理工具
- BEDtools: BED格式处理工具
- MACS2: 比对后找peak
- R: 统计作图
- Ngsplot: 可视化工具
- Deeptools: BAM文件分析工具, 可作图。
软件安装部分此处不介绍,毕竟如果你连软件安装都有困难,那你应该需要先学点Linux基础,或者去看生信必修课之软件安装
分析项目搭建
使用mkdir创建项目文件夹,用于存放后续分析的所用到的数据、中间文件和结果
mkdir -p r-chip/{analysis/0-raw-data,index,scripts,results}
个人习惯,在项目根目录下创建了四个文件夹
- analysis: 存放原始数据、中间文件
- index: 存放比对软件索引
- scripts: 存放分析中用到的脚本
- results: 存放可用于放在文章中的结果
后续所有的操作都默认在r-chip下进行,除非特别说明。
数据下载
根据文章提供的GEO编号(GEO: GSE97072)在NCBI上检索, 按照如下步骤获取该编号下所有数据的元信息, 我将其重命名为"download_table.txt"然后上传到服务器, 。
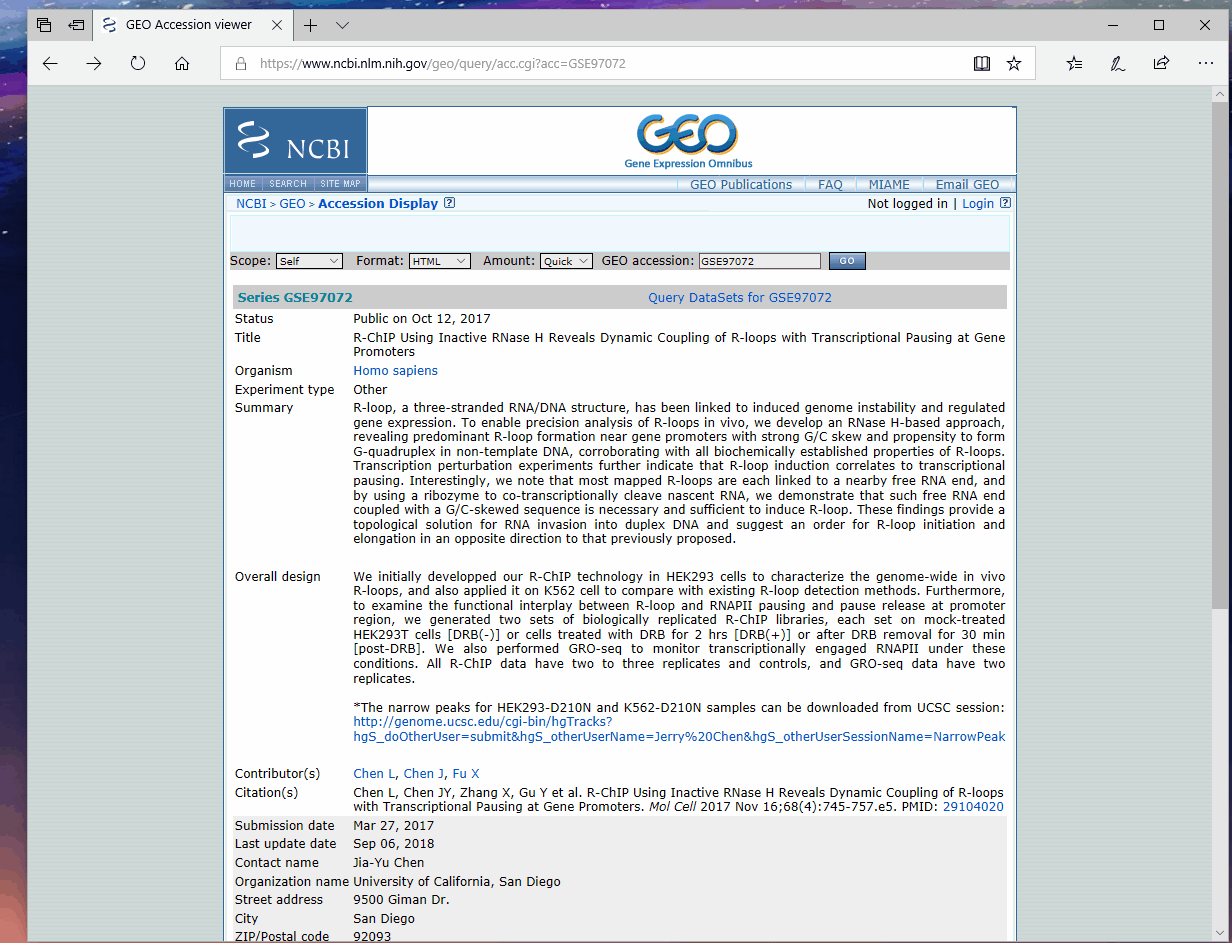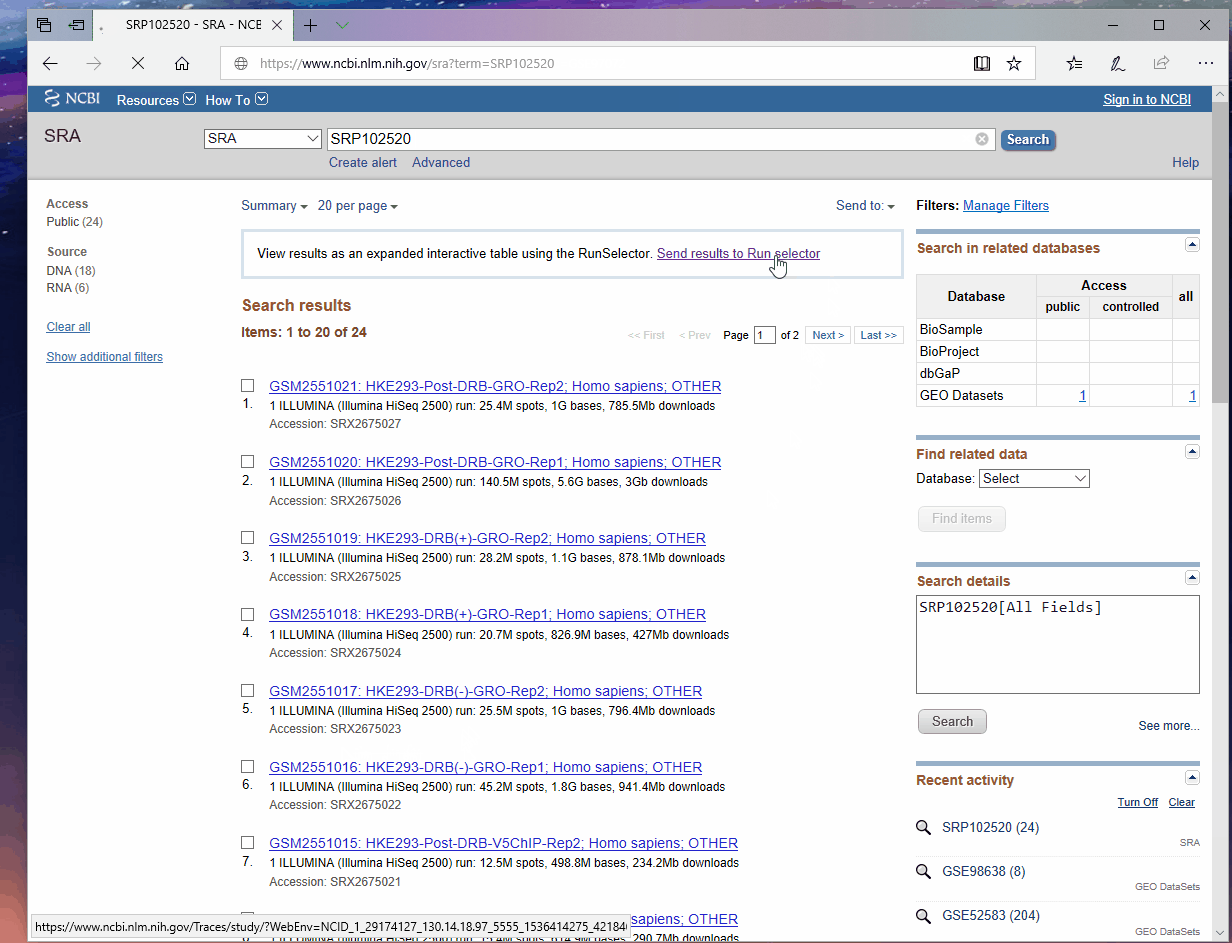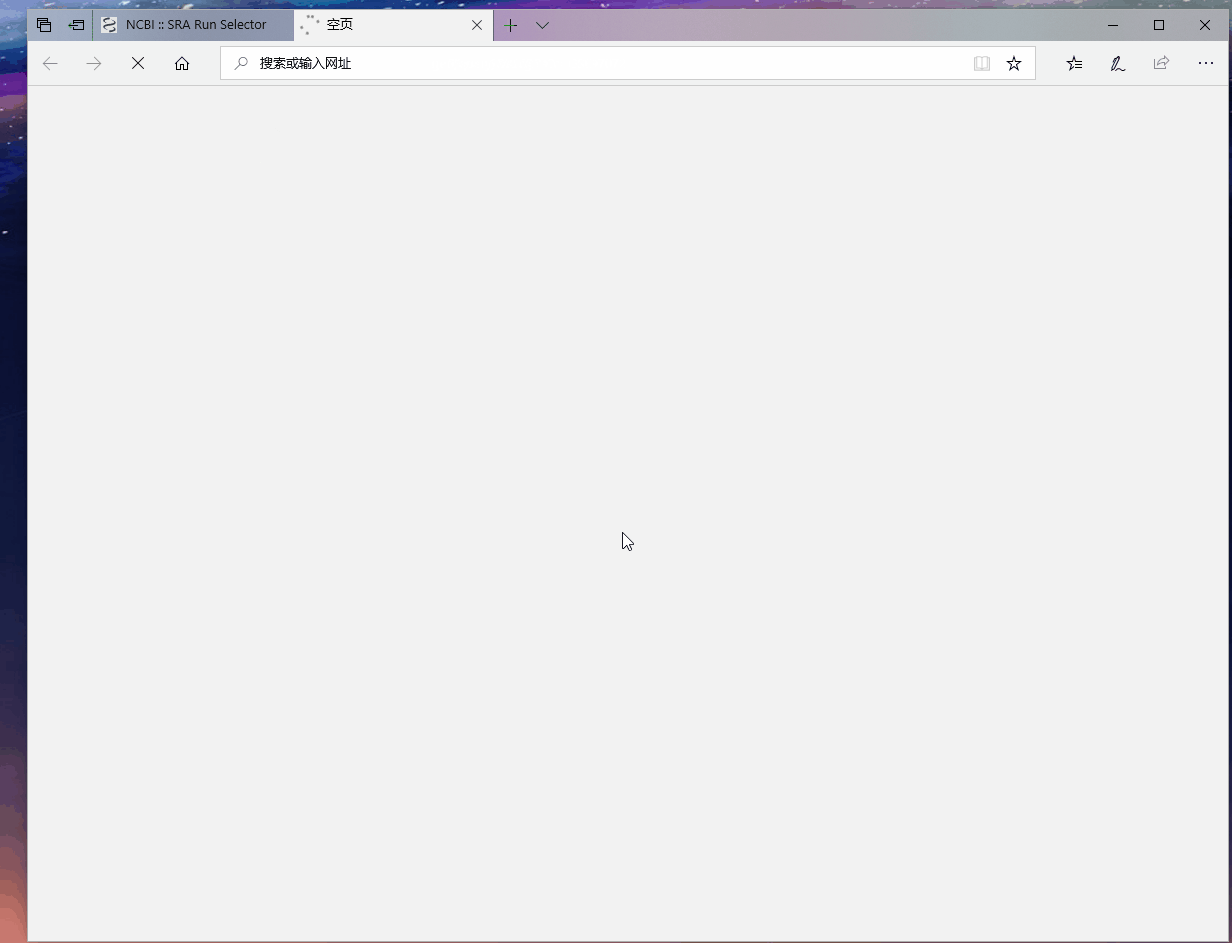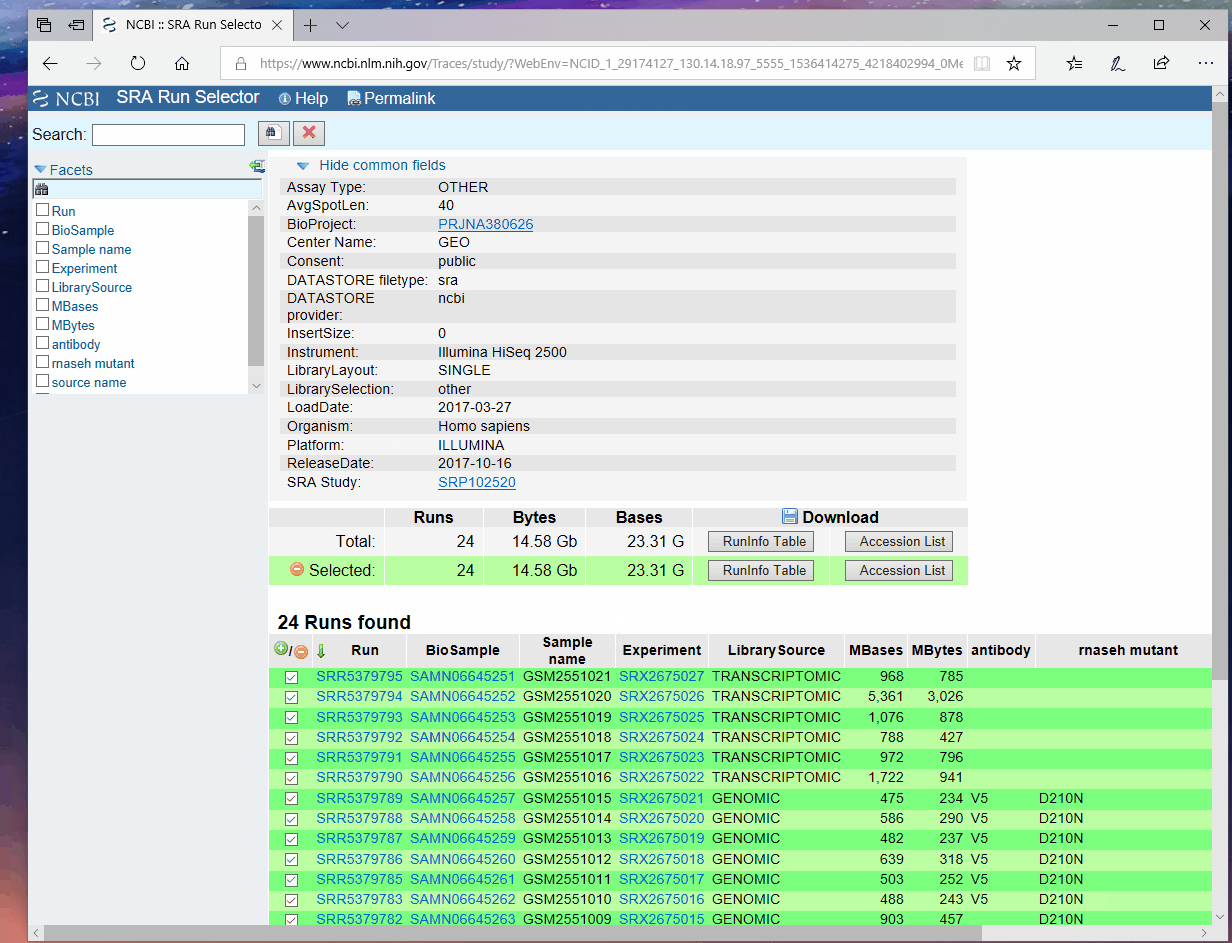
使用如下命令进行数据下载
tail -n+2 download_table.txt | cut -f 6 | xargs -i prefetch {} >> download.log &
下载的数据默认情况下存放在~/ncbi/public/sra, 需要用fastq-dump解压缩到analysis/0-raw-data. fastq-dump的使用说明见Fastq-dump: 一个神奇的软件
新建一个脚本,叫做uncompress.sh,存放在scripts文件下,代码如下
#!/bin/bash
set -e
set -o pipefail
set -u
tail -n+2 download_table.txt | cut -f 6 | while read id;
do
fastq-dump --gzip --split-3 --defline-qual '+' --defline-seq '@$ac-$si/$ri' &id -O analysis/0-raw-data &
done
然后用bash scripts/uncompress.sh运行。
注意:这是单端测序,所以每个SRR只会解压缩出一个文件
此外还需要下载human genome (hg19)的bowtie2索引,用于后续bowtie2比对。
curl -s ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/hg19.zip -o index/hg19.zip &
cd index
unzip hg19.zip