模式匹配中的正则表达式和k-mer
模式匹配指的是在看似杂乱无章的系统中找到符合要求的部分。比如说你想从基因组中寻找motif,转录因子结合位点,CDS, 或者检测测序结果里是否有接头等,这些行为都可以解读为根据已有的模式去寻找目标序列中符合要求的片段。后面介绍的序列模式匹配的两种方法,正则表达式和k-mers。
正则表达式的基础概念
正则表达式(regular expression)的概念,最初来自于20世纪40年代的两位神经学家(Warren McCulloch, Walter Pitts)研究神经元时提出的想法。后来数学家Stephen Kleene在代数学中正式描述了这种被他称之为“正则集合”的模型。并且,他还发明了一套简洁的方法表示正则集合,也就是正则表达式。
目前最快速的文本搜索工具grep就内置了正则表达式。grep起源于Unix中的ed编辑器的一条命令g/Regular Expression/p, 读作“Global Reular Expression Print”,即运用正则表达式的全局输出。由于这个功能太过实用,于是就从ed中独立出来,演变成了grep以及扩展版本的egrep。都知道grep因为有正则表达式所以很强大,但是正则表达式为何如此强大呢?
正则表达式的强大之处在于它是一套语法,分为两个部分,元字符(metacharacters)和普通文本字符(normal text characters)。
以语言类比,“我爱正则表达式”这句话可以抽象成“主谓宾”结构,主语是"我",谓语是"爱",宾语是“正则表达式”。这种语法还适用于其他语言,比如说英语就是"I love regular expression". 这种语法结构就是元字符,而构成句子的语言就是普通文字字符。
正则表达式的通用元字符
正则表达式有很多流派,不同流派之间的差异在于对元字符的支持程度。以下的元字符适用于GNU版本的grep, sed, awk. mac自带的是BSD版本。
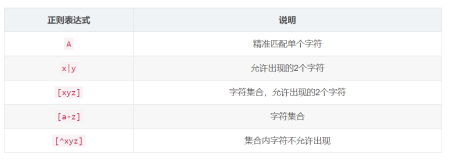
匹配单个字符的元字符:
| 元字符 | 匹配对象 |
|---|---|
| . | 匹配单个任意字符 |
| [...] | 匹配单个列出的字符 |
| [^...] | 匹配单个未列出的字符 |
| \char | 转义元字符成普通字符 |
提供计数功能的元字符:
| 元字符 | 匹配对象 |
|---|---|
| ? | 匹配0或1次 |
| * | 匹配0到n次 |
| + | 至少一次,最多不限 |
| {min,max} | 至少min次, 最多max次 |
匹配位置的元字符:
| 元字符 | 匹配对象 |
|---|---|
| ^ | 匹配一行的开头 |
| $ | 匹配一行的结尾 |
其他元字符:
| 元字符 | 匹配对象 |
|---|---|
| | | 匹配任意分割的表达式 |
| (...) | 限定多选结构的范围,标注量词的作用范围,为反向引用捕获元素 |
| \1, \2 | 反向引用元素 |
知道以上规则之后,推荐去http://regexpal.com/进行练习,以便更好的掌握。
使用正则表达式进行模式匹配
这一部分使用*nix系统自带的grep和Emboss工具集中的dreg,fuzznuc进行练习,所用练习数据为人类基因组的第22号染色体的DNA序列(fasta)以及高通量测序结果(fastq).
使用grep进行模式匹配
grep的强大之处它所做的事情就只有在文本搜索”正则表达式“定义的模式(pattern),如果找到就打印出来。可以使用man egrep查看所支持的参数。
egrep [options] pattern [file]
egrep [options] [-e pattern]... [-f FILE]... [FILE...]
# 参数参数
-e PATTERN: 定义多个模式
-f FILE: 从文本中读取模式
-w: 匹配整个单词
-v: 反向匹配
-i: 忽略大小写
-x: 仅仅选择整行匹配结果
-c: 计数
-n: 输出表明行号
-A/-B NUM: 同时输出后/前几行
注: grep有基础和扩展两个模式,基础模式支持的元字符较少,而egrep表示扩展的grep,支持的元字符较多。
利用grep搜索fastq文件中的序列:
# 获取fastq序列
fastq-dump --split-files SRR519926
# 寻找以ATG开头的序列
cat SRR519926_1.fastq | egrep "^ATG" --color=always | head
# 寻找以ATG结尾的序列
cat SRR519926_1.fastq | egrep "ATG\$" --color=always | head
# 寻找TAATA或TATTA
cat SRR519926_1.fastq | egrep "TA[A,T]TA" --color=always | head
# 寻找TAAATA或TACCTA
cat SRR519926_1.fastq | egrep "TA(AA|CC)TA" --color=always | head
## 比较[]和(|)的区别
# 寻找TA后面多个A或没有,随后是TA的序列
cat SRR519926_1.fastq | egrep "TA(A*)TA" --color=always | head
# 寻找TA后面接着1个以上A,之后是TA的序列
cat SRR519926_1.fastq | egrep "TA(A+)TA" --color=always | head
# 寻找TA后面跟着2到5个的序列,随后是TA
cat SRR519926_1.fastq | egrep "TAA{2,5}TA" --color=always | head
# 找找illumina的接头序列AGATCGG
cat SRR519926_1.fastq | egrep "AGATCGG.*" --color=always | head
使用dreg和fuzznuc对核酸数据进行匹配
一般而言,对于fastq文件,*nix提供的grep基本已经够用了。但是对于fasta文件而言,可能会存在一些问题,比如说下面这段序列,你是绝对匹配不到TAATA,尽管你用肉眼都能看的出来,TAA后面就是TA,但是grep就是找不到。
## 22号染色体数据
curl http://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/chr22.fa.gz | gunzip > chr22.fa
## 查看序列
tail -n 1000 chr22.fa | head -n 5
CTGCCTGCGGGGAGGGGGTGGGGAAGGTGTTAATGATGCTGATCCCTACT
TCTGCTTCAAGGAGATCTGGTGGGGAATTCTTCCACCAGTCCAGAGTTTG
CTGGTGCTGACCTCATCCCTGTATCACGGGCCTAGAATGTGGGAGGCTAA
TAGGATGGGTGGGTTGCAGGAGGTAGAAGAGGGGATGGCCTAGAGAGTTT
CTCCATTCAGAGCTGGAGAGTTGTTGAAGGGAAGGGTATTTTAAAAGGGC
tail -n 1000 char22.fa | head -n5 | grep TAATA
真相只有一个,这段序列为了显示方便,存放的时候被折叠了,所以TAA和TA之间其实存在一个换行符。解决方法有两个,第一个是去掉序列中的换行符tail -n 1000 chr22.fa | head -n 20 | tr -d '\n' | egrep -o -i 'TAATA',第二个就是使用emboss工具集的dreg或fuzznuc。
# 安装emboss工具集
conda install -c bioconda emboss # for linux
brew install emboss # for Macos
# 使用方法见官方说明
# http://emboss.sourceforge.net/apps/cvs/emboss/apps/fuzznuc.html
# http://emboss.sourceforge.net/apps/cvs/emboss/apps/dreg.html
dreg和funzznuc功能基本上一摸一样,使用方法也是几乎一样,但是dreg比funzznuc支持的正则表达式多,因此通常使用dreg。
# 首先去找之前的TAATA
tail -n 1000 chr22.fa | head -n 5 | dreg -filter -pattern 'TAATA'
# 寻找TTAGGG重复的序列
cat chr22.fa | dreg -filter -pattern '(TTAGG){20,30}'
dreg的用法和grep基本上一致的,这里不再赘述。对于氨基酸序列和翻译后的蛋白序列,则有preg和fuzzprt,fuzztran。
K-Mers
k-mer指的是一个字符串中所有长度为k的子字符串的集合。如ATCGA的所有k-mer如下:
- 2-mers:
AT,TG,GCandCA - 3-mers:
ATG,TGCandGCA - 4-mers:
ATGC,TGCA - 5-mers:
ATGCA
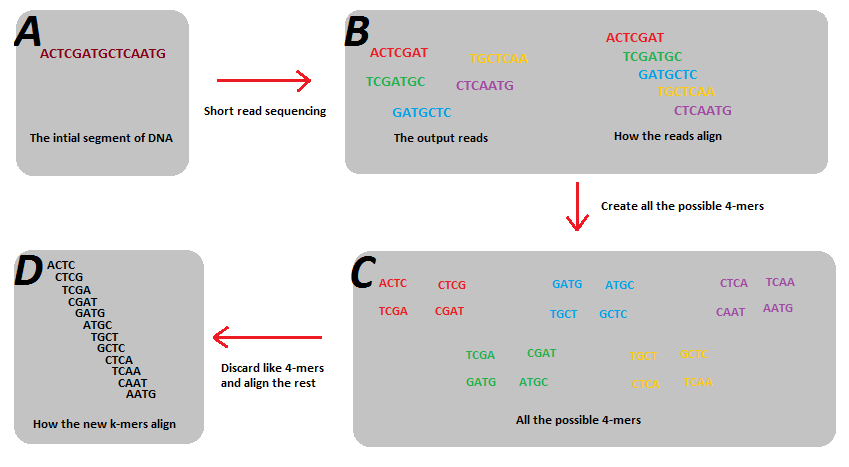
k-mers的主要使用场景是基因组组装,如下图所示
当然他还能用于:
- 纠错:那些稀有不常见的k-mers,可能仅仅是测序错误.
- 分类:基因组中特异性的k-mers可以用来区分不同物种。 Classification: certain k-mers may uniquely identify genomes.
- Pseudo-alignment(伪比对): 目前RNA-Seq定量分析中出现了一类alignment-free工具,其原理就是先准备不同基因的k-mers的索引,通过将read的k-mers和k-mers索引比较,从而对基因进行计数。
案例一: 使用jellyfish统计k-mers
# 安装jellyfish
conda install -c bioconda jellyfish # for linux
brew install jellyfish # for MacOS
# 获取序列数据
efetch -id KU182908 -db nucleotide -format fasta > KU182908.fa
# 对fasta里面的k-mers计数,-C表示双端, -m表示k的长度,-s10M表示初始hash为10M,根据数据量大小而定。
jellyfish count -C -m 10 -s10M KU182908.fa
# k-mers 频次展示
jellyfish histo mer_counts.jf
#1 17736
#2 539
#3 26
#4 6
#6 1
#7 4
# 出现7次的k-mers
jellyfish dump -L 7 mer_counts.jf
>7
ATGAAGATTA
>7
TTAAGAAAAA
>7
AATCTTCATC
>7
ATTAAGAAAA
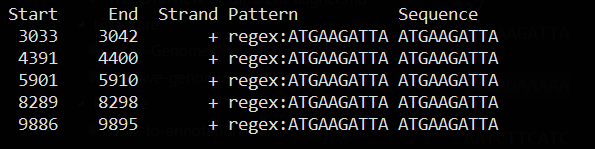
根据出现7次10-mers序列对原来序列进行模式匹配
cat KU182908.fa | dreg -filter -pattern ATGAAGATTA
案例二:利用k-mers估计基因组
这一部分内容属于基因组survey的一部分。在组装基因组前需要对基因组有一个评估,比如说杂合率,重复率,基因组大小等内容,这些就需要用到jellyfish。
# 自行找双端测序的测试文件, 假设是A_1.fq.gz和A_2.fq.gz
# 数据的19-mer计数
jellyfish count -m 19 -s 300M -t 4 -C <(zcat A_1.fq.gz) <(zcat A_2.fq.gz)
jellyfish histo mer_counts.jf > counts.txt
# 用R作图
histo <- read.delim("counts.txt", sep = " ",header = FALSE)
plot(histo[3:200,], type="l")
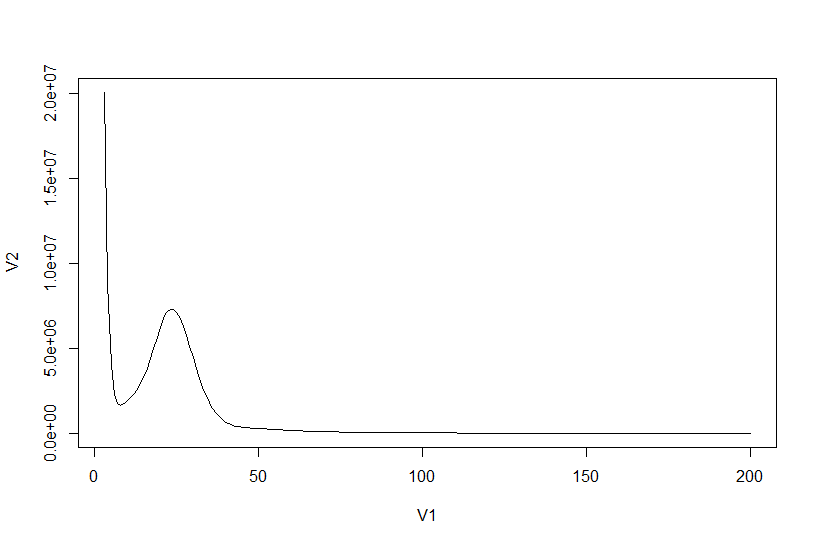
根据上图找到单峰区间,并计算总k-mers数
plot(histo[3:200,], type="l")
points(histo[6:50,]) # 根据点图确认单峰区间在6到50
sum(as.numeric(histo[3:9597,1] * histo[3:9597,2])) # 5477901244
确定峰所在位置,并且计算基因组大小
histo[6:50,] # 24
sum(as.numeric(histo[3:9597,1] * histo[3:9597,2])) / 24 / 1000 /1000 # 约为 208M
参考资料: https://bioinformatics.uconn.edu/genome-size-estimation-tutorial/
一些更加方便的脚本工具:
- estimate_genome_size.pl: https://github.com/josephryan/estimate_genome_size.pl
- KmerGenie: http://kmergenie.bx.psu.edu/
- 华大的GCE: ftp://ftp.genomics.org.cn/pub/gce
- ALLPATHS-LG的 findErrors模块。
最后还有一个问题:FastQC的结果里面也有一个k-mers,应该如何看待呢?答:一笑而已,请用jellyfish统计